实验四介观动力学模拟
- 格式:doc
- 大小:1.78 MB
- 文档页数:15

化学反应动力学模型的建立与分析一、化学反应动力学模型建立的步骤1.收集实验数据:首先需要进行实验,收集反应物浓度随时间变化的数据。
实验条件可以根据需要进行调节,如温度、浓度、催化剂等。
可以通过实验方法,如光谱、色谱、电位法、质谱等对反应物浓度进行测定。
2. 确定反应级数:根据实验数据分析反应级数。
一级反应速率与反应物浓度成正比,二级反应速率与反应物浓度的平方成正比。
可以通过对反应物浓度进行对数处理,绘制ln[A] vs t或者1/[A] vs t得到指数趋势图,从图形上判断反应级数。
3.构建速率方程:根据反应级数确定速率方程。
一级反应速率方程为r=k[A],二级反应速率方程为r=k[A]²。
可以将速率方程与实验数据进行拟合,确定反应速率常数k。
4.推导反应机理:通过实验数据和速率方程,推导反应机理。
可以根据速率方程,确定反应的中间产物和反应活化能。
二、化学反应动力学模型分析的方法1.速率常数的求解:速率常数k是化学反应速率与反应物浓度的关系参数,可以通过实验数据拟合得到。
常见的拟合方法有线性拟合和非线性拟合。
可以通过最小二乘法拟合实验数据点,得到速率常数的估计值。
2.动力学模型的验证:建立化学反应动力学模型后,需要进行模型验证。
可以使用其他实验数据进行验证,或者通过计算机模拟进行验证。
验证的目的是确认模型的准确性和可靠性,以便用于预测和解释其他化学反应。
3.动力学参数的确定:根据实验数据和模型分析结果,确定动力学参数,如反应速率常数k、反应级数、反应活化能等。
这些参数可以提供反应机制、反应速率常数以及温度、压力等条件的依赖关系。
4.动力学模型的应用:通过分析动力学模型,可用于预测反应速率、优化反应条件、设计反应工艺和控制反应过程。
通过改变温度、浓度、催化剂等条件,可以实现改变反应速率和反应选择性的目的。
总结:建立和分析化学反应动力学模型是研究反应速率和机理的重要方法。
通过收集实验数据,确定反应级数和速率方程,推导反应机理,可以建立动力学模型。

分子动力学模拟实验步骤
嘿,朋友们!今天咱来聊聊分子动力学模拟实验步骤这档子事儿。
咱先得有个明确的目标吧,就好比你要去个地方,得知道去哪儿呀!这分子动力学模拟实验也一样,你得清楚自己要研究啥。
然后呢,就是选个合适的模型啦。
这就像你出门得挑双合脚的鞋子,模型不对,那后面的路可就不好走咯。
得仔细琢磨琢磨,找个能准确反映实际情况的模型。
接下来,设置好各种参数。
这可不能马虎,就跟你调电视音量似的,得恰到好处。
温度啊、压力啊、粒子间的相互作用啥的,都得考虑周全。
再之后,让计算机开始运算吧!这计算机就像个勤劳的小蜜蜂,嗡嗡嗡地帮咱干活。
咱就等着看它给出的结果。
在这过程中,你得时刻盯着点,看看有没有啥不对劲的地方。
这就好比你煮汤的时候得时不时看看火,别煮糊了呀。
等计算机算完了,就该分析结果啦。
这可需要点真本事,得从那些密密麻麻的数据里找出有用的信息。
这就像在一堆沙子里找金子,得有耐心,还得有好眼神。
分析完结果,要是不满意咋办?那就重新来呗!别灰心,科学家们不都是这样一点点摸索过来的嘛。
你说这分子动力学模拟实验像不像搭积木?一块一块地往上搭,搭错了就重新来,直到搭出你想要的那个城堡。
这中间的乐趣和挑战,只有试过才知道呀!
总之,分子动力学模拟实验可没那么简单,但也绝对不是高不可攀的。
只要咱一步一个脚印,认真去做,肯定能发现其中的奥秘。
大家加油干吧,说不定下一个重大发现就是你做出来的呢!
原创不易,请尊重原创,谢谢!。

化学反应动力学模型建立与模拟方法教程为了深入了解化学反应过程并预测其动力学行为,建立适当的模型是至关重要的。
化学反应动力学模型的建立能够提供相关参数和反应速率方程,从而使研究者能够更好地理解反应机理和优化反应条件。
本文将介绍化学反应动力学模型建立与模拟的基本步骤和方法。
第一步是数据收集。
在建立反应动力学模型之前,需要收集实验数据。
实验数据应包含反应物浓度、反应温度和反应速率等信息。
这些数据可通过实验室实验或文献调研获得。
数据收集是建立可靠模型的基础,因此务必确保数据的准确性和全面性。
第二步是选择适当的模型。
根据反应的特性和目的,可以选择不同类型的动力学模型。
常见的模型包括一级反应、二级反应、阻滞动力学和微分动力学等。
选取合适的模型需要考虑反应机理和反应条件等因素。
第三步是参数估计。
参数估计是建立反应动力学模型过程中的关键步骤。
参数包括反应速率常数、活化能和反应级数等。
参数估计可以通过不同的统计方法、优化算法和曲线拟合等进行。
最常用的方法是最小二乘法,通过最小化实验数据和模型预测值之间的平方误差来估计参数。
第四步是模型验证。
模型验证是确认所建立模型的可靠性和准确性的重要环节。
通过将模型的预测结果与实验数据进行比较,可以评估模型的精确度和适用性。
如果模型表现良好,即能够准确预测实验数据,则可以认为该模型是可靠的。
除了建立动力学模型,还可以利用模型进行反应模拟。
反应模拟可以预测不同反应条件下的反应动力学行为。
通过改变反应物浓度、温度和反应时间等参数,可以预测反应速率和产物生成量的变化趋势。
反应模拟能够帮助研究者优化反应条件,提高反应效率和选择合适的反应条件。
在进行反应模拟之前,需要对模型进行合理的参数选择。
参数选择可以基于实验数据或文献中已有的参数值。
同时,也可以通过灵敏度分析来评估参数对模型预测结果的影响程度。
灵敏度分析可以帮助确定关键参数,进一步优化模型。
反应模拟的过程中,可以使用不同的数值方法求解动力学方程。

分子动力学模拟实验报告篇一:分子动力学实验报告 md2分子动力学实验报告( XX 至 XX 学年第_2_学期)班级:姓名:学号:实验名称:晶体点缺陷成绩:一、实验目的计算空位形成能和间隙原子形成能。
探究形成的空位和间隙原子所在的位置不同其形成能的变化。
以及空位和间隙原子的浓度不同时其空位能和间隙原子形成能的变化。
二、实验原理点缺陷普遍存在于晶体材料中,它是晶体中最基本的结构缺陷,对材料的物理和化学性质影响很大。
根据点缺陷相对于理想晶格位置可能出现的几种主要偏差状态,可将其命名如下:(1)空位:正常节点位置上出现的原子空缺。
(2)间隙原子(离子):指原子(离子)进入正常格点位置之间的间隙位(本文来自:小草范文网:分子动力学模拟实验报告)置。
(3)杂质原子(离子):晶体组分意外的原子进入晶格中即为杂质,杂质原子若取代晶体中正常格点位置上的原子(离子)即为置换原子(离子),也可进入正常格点位置之间的间隙位置而成为填隙的杂质原子(离子)。
一般情况下,空位、间隙原子都是构成晶体的原子或离子偏离原有格点所形成的热缺陷。
在一定温度下,晶体中各原子的热振动状态和能量并不同,遵循麦克斯韦分布规律。
热振动的原子某一瞬间可能获得较大的能量,这些较高能量的原子可以挣脱周围质点的作用而离开平衡位置,进入到晶格内的其他位置,于是在原来的平衡格点位置上留下空位。
根据原子进入晶格内的不同位置,可以将缺陷分为弗伦克尔(Frenkel)缺陷和肖特基(Schottky)缺陷。
点缺陷都只有一个原子大小的尺度,因此不容易通过实验对其进行直接的观察。
而且实验方法研究缺陷时利用较多的还是缺陷对晶体性质的影响。
例如,通过测量晶体的膨胀率和电阻率的变化规律,即可对点缺陷的存在、运动和相互作用等方面展开间接的研究。
分子动力学方法对金属材料原子尺度物理和化学过程的研究具有实验法无法比拟的优势,可直观的模拟和分析晶体中的点缺陷。
若我们搭建完整晶体的原子个数为N,能量为E1,通过删除和增加一个原子得到空位和间隙原子,充分弛豫后体系能量为E2,则空位形成能Ev 和间隙原子形成能Ei分别为:三、实验过程(1)进入2_point文件夹$cd口2_point(2)运行in.inter文件,得到Cu的八面体间隙原子的图像,以及体系的总能量的变化,计算出八面体间隙原子的形成能。

化学反应的动力学模拟化学反应中的动力学过程是指反应速率的变化规律,即反应物转化为生成物的速率。
为了更好地理解和研究化学反应的动力学规律,科学家们发展了许多动力学模拟方法。
本文将介绍一些常用的化学反应动力学模拟技术及其应用。
一、量子力学方法量子力学是一种精确描述原子和分子行为的数学理论。
在化学反应动力学模拟中,研究人员通常使用量子力学方法,如密度泛函理论(DFT)和分子力场(MM)等。
DFT可以模拟物质的电子结构和分子振动,通过计算反应物和过渡态的能量,可以得到反应的活化能、反应速率常数等动力学参数。
分子力场方法则是通过精确描述原子间相互作用势能曲线,进而计算反应的转化率和反应速率。
二、分子动力学模拟方法分子动力学模拟是一种基于牛顿力学原理的计算方法,通过模拟原子和分子的运动轨迹,研究它们在不同温度和压力下的行为。
分子动力学模拟可以计算反应物和过渡态的构象、能量、速度等信息,从而预测反应的速率常数和反应机理。
三、反应坐标方法反应坐标方法是一种通过构建反应的势能面来模拟化学反应动力学的方法。
在反应坐标方法中,将反应转化为一系列过渡态,通过计算过渡态的活化能、反应路径等参数,可以探究反应的速率和反应机理。
常用的反应坐标方法有能均面动力学和内禀反应坐标。
四、蒙特卡洛模拟方法蒙特卡洛模拟是一种随机模拟方法,通过大量的概率采样,模拟系统的演化过程。
在化学反应的动力学模拟中,蒙特卡洛模拟能够模拟反应物和过渡态的构象空间,通过大量的随机采样,可以得到反应的速率和转化率等动力学参数。
五、应用案例动力学模拟在化学反应中有广泛的应用。
例如,科学家通过动力学模拟探究了一系列重要反应的速率常数和反应机理。
同时,动力学模拟还可以帮助研究人员优化反应条件,提高反应的速率和选择性。
总结:化学反应的动力学模拟方法包括量子力学方法、分子动力学模拟、反应坐标方法和蒙特卡洛模拟等。
这些方法能够帮助我们理解反应的速率和机理,优化反应条件,推动化学反应领域的研究和应用。

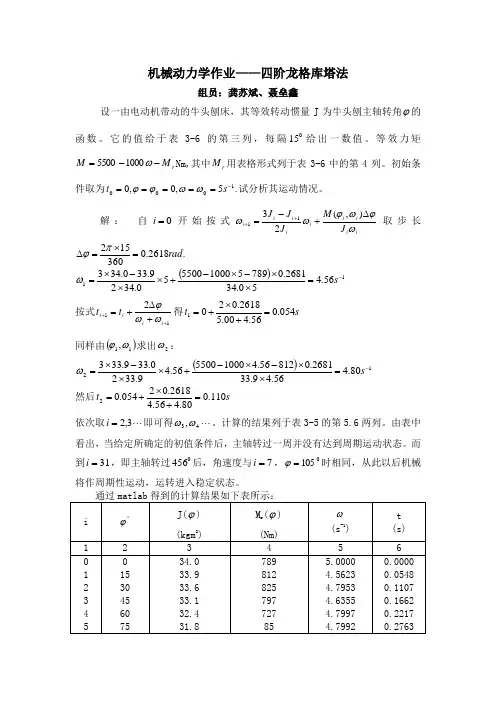
机械动力学作业——四阶龙格库塔法组员:龚苏斌、聂垒鑫设一由电动机带动的牛头刨床,其等效转动惯量J 为牛头刨主轴转角ϕ的函数。
它的值给于表3-6的第三列,每隔015给出一数值。
等效力矩r M M --=ω10005500Nm,其中r M 用表格形式列于表3-6中的第4列。
初始条件取为.5,0,01000-=====s t ωωϕϕ试分析其运动情况。
解: 自0=i 开始按式ii i i i i i i i J M J J J ωϕωϕωω∆+-=++),(2311取步长.2618.0360152rad =⨯=∆πϕ ()1156.450.342681.078951000550050.3429.330.343-=⨯⨯-⨯-+⨯⨯-⨯=s ω 按式112+++∆+=i i i i t t ωωϕ得s t 054.056.400.52618.0201=+⨯+=同样由()11,ωϕ求出2ω:()1280.456.49.332681.081256.41000550056.49.3320.339.333-=⨯⨯-⨯-+⨯⨯-⨯=s ω 然后s t 110.080.456.42618.02054.02=+⨯+=依次取 3,2=i 即可得 43,ωω。
计算的结果列于表3-5的第5.6两列。
由表中看出,当给定所确定的初值条件后,主轴转过一周并没有达到周期运动状态。
而到31=i ,即主轴转过0456后,角速度与7=i ,0105=ϕ时相同,从此以后机械将作周期性运动,运转进入稳定状态。
运用四阶龙格—库塔公式)22(6143211k k k k i i ++++=+ωω其中,),(1i i hf k ωϕ=、)2,2(12k h hf k i i ++=ωϕ、)2,2(23k hhf k i i ++=ωϕ、),(33k h hf k i i ++=ωϕ。
主要函数ωϕωωϕωϕJ d dJM f 2),(),(2-=,其中ϕϕ∆-=+i i i J J d dJ 1)(利用上述的公式在matlab 中编写程序,主要就是编写循环函数。

动力学模拟的主要步骤那动力学模拟呢,一开始得确定模拟的系统。
这就像是你要搭建一个小世界,你得先想好这个世界里都有啥呀。
比如说,是一堆原子呢,还是一些分子,得把这些参与的“小成员”都确定好。
这一步可不能马虎哦,要是少了哪个关键的“小成员”,那后面模拟出来的结果可能就会差之千里啦。
接着呢,要给这个系统设定初始条件。
这就好比给这个小世界一个初始的状态。
比如这些原子或者分子的初始位置、初始速度啥的。
你想啊,如果一开始就乱套了,那这个小世界的发展肯定也不正常呀。
就像玩游戏,开局的布局和状态要是不对,后面可就难搞喽。
然后呢,就是选择合适的力场啦。
力场就像是这个小世界里的规则制定者。
它决定了这些原子或者分子之间是怎么相互作用的。
不同的力场适用于不同的系统哦。
选错了力场,就像是在足球比赛里用了篮球的规则,那整个游戏就乱套啦。
再之后就是进行模拟计算啦。
这个时候计算机就开始忙乎起来啦,根据前面设定的那些条件和规则,开始计算这个系统随着时间的推移会发生什么样的变化。
这就像是看着这个小世界慢慢发展,看原子们怎么跑来跑去,分子们怎么相互碰撞、结合或者分开。
这个过程可能会有点漫长呢,就像等一朵花开一样,要有耐心。
最后呀,得对模拟的结果进行分析。
这就像是看完一场电影后,要总结一下电影讲了啥一样。
看看模拟出来的数据是不是符合我们的预期,有没有发现什么新的有趣的现象。
要是结果不太对,那可能就得回头看看前面的步骤是不是哪里出了岔子。
动力学模拟虽然有点复杂,但就像一场有趣的探索之旅呢。
每一步都很重要,就像拼图一样,少了一块都拼不出完整的画面。
宝子,这么一讲是不是感觉也没那么神秘啦?。

《计算材料学》实验讲义粗粒度模拟实验名称:介观动力学模拟一、前言1、介观模拟简介长期以来,化学家致力于从分子水平研究物质及其变化,而化学工程工作者主要研究物质在宏观体系的行为,介观层次的化学正是联系微观及宏观的桥梁,是从分子到材料的必由之路,同生命过程也有密切的关联。
由于介观模拟能够模拟的空间尺度(纳米到微米)、时间尺度(纳秒到微妙)更大,应用介观模拟方法可以模拟更加复杂的体系,例如:高分子熔体,高分子稀溶液自组装,表面活性剂溶液自组装,磷脂膜等胶体化学,高分子,生物大分子相关的内容。
目前介观模拟的方法很多,例如耗散颗粒动力学模拟方法(dissipative particle dynamics,DPD),它是根据Hoogerbrugge和Koelman提出的一种针对柔性(soft)球模型流体动力学的模拟,并通过引入粒子间的谐振动势,来模拟聚合物的性质;元胞动力学方法(CDS),基于重整化群理论,对时间相关的Ginzburg-Landau方程直接用数值计算的方法在离散空间上进行描述。
其中单个元胞的演化通常用双曲正切函数表示;动态密度泛函方法(DDFT或MesoDyn),应用于高分子体系,建立在粗粒化高斯链模型的基础上,实际上是一个动态的自洽场方法,使用了朗之万方程(Langevin’s equation)来描述体系演化的动力学。
(1)MS-Mesocite简介MS Mesocite是一个基于粗粒度模拟方法的、可以对广泛体系进行模拟研究的分子力学工具集,模拟的对象大小尺寸在纳米到微米尺度范围,相应地,模拟变化的时间范围落在纳秒至微秒区间。
MS Mesocite的模拟对象遍及多种工业领域,比如复合材料、涂料、化妆品以及药物控缓释等,它可以提供流体在平衡态下、在有剪切力存在下以及其它受限制条件下的结构与动力学性质。
MS Mesocite 的突出特点是使用完全区别于传统介观模拟技术,转而采用力场(Forcefield)方法—比如MS Martini力场—来描述粗粒度之间的相互作用,从而得到体系的结构、和动力学特性,分析函数主要有角度分布,密度分布,径向分布函数,二面角分布,均方根位移等。

化学反应中的动力学分析与模拟第一章:引言化学反应是化学学科的核心之一,它涉及到许多重要的现象和过程。
了解化学反应机理和动力学是化学领域的重要研究方向之一。
通过动力学分析和模拟,可以更好地理解化学反应的动态过程和反应规律,为化学反应的设计和优化提供指导。
本文旨在介绍化学反应动力学分析和模拟的基本概念,探讨重要的化学反应动力学模型,并评估现代计算机模拟方法在化学反应动力学研究中的应用。
第二章:化学动力学基础化学反应动力学研究的基础是化学反应动力学方程。
这些方程描述了反应物质在不同温度、压力、浓度或电场等条件下发生反应的速率。
反应速率的确定需要考虑以下因素:反应物浓度、反应物种类、反应条件(如温度和压力)和催化剂。
在化学领域,最常见的化学反应动力学方程形式为速率常数表达式。
速率常数k是描述反应速率的重要物理量,它与反应物的浓度、反应物种类和温度等因素有关。
在理论研究中,k通常由反应物分子之间碰撞的频率和能量来确定。
第三章:反应动力学模型化学预测模型是化学反应动力学模拟研究的核心。
反应动力学模型可以帮助研究人员更好地理解反应机理和预测反应成果。
下面介绍几种化学反应动力学模型:1. 一阶反应模型一阶反应模型是最常用的动力学模型之一。
在一阶反应中,反应物分子以自身浓度及速率常数k为基础反应。
一阶反应模型对于不同化学反应涉及了种类相似的动态过程,如放射性射线的衰变和许多金属腐蚀。
2. 二阶反应模型二阶反应模型是描述许多化学反应的动力学模型。
在这种模型中,两个反应物分子交换电子,生成新的化合物。
二阶反应模型通常涉及带电离,酸碱反应和许多重要的生化反应。
3. 多级反应模型多级反应模型常用于描述极其复杂的反应动态。
在多级反应中,一种化学物质的生成需要通过多个中间步骤完成。
多级反应模型能够更好地描述反应动态和机理。
第四章:化学反应动力学的模拟计算化学方法已经成为现代化学科学中的重要组成部分。
通过计算机模拟,可以对复杂的化学反应机理和动态等进行定量研究。

实验报告化学实验中的反应动力学实验报告:化学实验中的反应动力学1. 引言反应动力学是化学研究中的重要领域,研究化学反应速率随时间的变化规律。
实验方法是探究反应动力学的重要手段之一。
本实验旨在通过测量不同条件下化学反应速率的变化,探索反应速率与反应物浓度、温度等因素之间的关系。
2. 实验目的本实验的目的是通过观察酶催化反应在不同温度和底物浓度下的变化规律,探究反应速率与温度、浓度之间的关系。
3. 实验设计3.1 实验材料和仪器- 酶溶液- 底物溶液- 温水浴- 容量瓶- 比色皿- 分光光度计3.2 实验步骤1. 准备不同浓度的底物溶液,分别标注。
2. 在比色皿中加入一定量的酶溶液。
3. 将比色皿放入分光光度计中,记录吸光度初始值。
4. 向比色皿中加入底物溶液,立即开始计时。
5. 每隔一段时间,记录吸光度的变化。
6. 重复上述步骤,但在不同温度下进行实验。
4. 结果与讨论4.1 实验结果根据实验数据,我们可以得出以下结果:随着底物浓度的增加,反应速率呈正相关关系;随着温度的升高,反应速率也增加。
4.2 结果分析根据我们的实验结果,我们可以发现反应速率与底物浓度和温度有关。
随着底物浓度的增加,反应物的有效碰撞频率增加,从而增加了化学反应的速率。
而温度的升高则加快了反应物的动能,进一步促进了反应速率的增加。
此外,我们还可以通过测量反应速率随时间的变化曲线,得出反应级数和速率常数等动力学参数,从而更加全面地了解反应动力学规律。
5. 结论本实验通过测量酶催化反应在不同底物浓度和温度下的速率变化,研究了反应动力学的基本规律。
实验结果表明底物浓度和温度对反应速率具有显著影响。
进一步研究反应动力学以及相关因素的变化规律,对于理解化学反应的本质和优化反应条件具有重要意义。
水溶液的介观结构与形成机理虚拟仿真实验实验报告(一)水溶液的介观结构与形成机理虚拟仿真实验实验报告作为一名资深的创作者,我参与了一项有关“水溶液的介观结构与形成机理”的虚拟仿真实验。
以下是我的实验报告:实验目的通过虚拟仿真实验,探究水溶液的介观结构和形成机理,深入了解水分子之间的相互作用力和溶质分子在水溶液中的行为。
实验内容1.创建模拟环境:使用LAMMPS软件建立水溶液的模拟环境,并设定合适的模拟时间和温度。
2.模拟分析:使用VMD软件对模拟结果进行可视化分析,了解水分子的分布和运动情况。
3.数据处理:使用Origin软件对模拟数据进行处理和分析,如可视化绘制分子轨迹、径向分布函数等。
实验结果1.水分子的分布:通过VMD软件观察发现,水分子围绕着溶质分子形成水合壳,形成一定的结构化网络。
2.水分子的运动:水分子在水溶液中的运动方式比较复杂,既有碰撞,又有相互作用力的作用,影响其运动轨迹和速度。
3.溶质分子的行为:溶质分子具有特定的亲疏水性质,在水溶液中可能会发生溶解、离解、水合等行为。
实验结论通过本次虚拟仿真实验,我们深入了解了水溶液介观结构的形成机理及其影响因素,水分子之间相互作用力和溶质分子的行为规律。
这对于我们更好地理解水和溶液的性质、探索化学与生命科学等领域的研究具有重要的意义。
实验优缺点优点1.虚拟仿真实验提供了一种安全、经济、高效的实验方法,避免了实验中可能会出现的安全风险和高成本的实验费用。
2.虚拟仿真实验可以帮助我们更好地了解和掌握真实实验的基本原理和实验流程,提高实验操作技能和实验设计能力。
3.虚拟仿真实验促进了实验教学方法的创新,更加注重学生的实践和体验,激发了学生的兴趣和学习动力。
缺点1.虚拟仿真实验同真实实验相比存在着一定的模型简化和误差不确定性的问题,可能会影响实验的准确性和可靠性。
2.虚拟仿真实验可能会缺少一些真实实验所具有的情境感、主观感受等因素,不利于学生对实验过程的深入思考和综合评价。
实验四:介观动力学模拟_27396《计算材料学》实验讲义实验八:介观动力学模拟一、前言1、介观模拟简介长期以来,化学家致力于从分子水平研究物质及其变化,而化学工程工作者主要研究物质在宏观体系的行为,介观层次的化学正是联系微观及宏观的桥梁,是从分子到材料的必由之路,同生命过程也有密切的关联。
由于介观模拟能够模拟的空间尺度(纳米到微米)、时间尺度(纳秒到微秒)更大,应用介观模拟方法可以模拟更加复杂的体系,例如:高分子熔体,高分子稀溶液自组装,表面活性剂溶液自组装,磷脂膜等胶体化学,高分子,生物大分子相关的内容。
目前介观模拟的方法很多,例如耗散颗粒动力学模拟方法(dissipative particle dynamics,DPD),它是根据Hoogerbrugge和Koelman提出的一种针对柔性(soft)球模型流体动力学的模拟,并通过引入粒子间的谐振动势,来模拟聚合物的性质;元胞动力学方法(CDS),基于重整化群理论,对时间相关的Ginzburg-Landau方程直接用数值计算的方法在离散空间上进行描述。
其中单个元胞的演化通常用双曲正切函数表示;动态密度泛函方法(DDFT或MesoDyn),应用于高分子体系,建立在粗粒化高斯链模型的基础上,实际上是一个动态的自洽场方法,使用了朗之万方程(Langevin’s equation)来描述体系演化的动力学。
(1)MS-Mesocite简介MS Mesocite是一个基于粗粒度模拟方法的、可以对广泛体系进行模拟研究的分子力学工具集,模拟的对象大小尺寸在纳米到微米尺度范围,相应地,模拟变化的时间范围落在纳秒至微秒区间。
MS Mesocite的模拟对象遍及多种工业领域,比如复合材料、涂料、化妆品以及药物控缓释等,它可以提供流体在平衡态下、在有剪切力存在下以及其它受限制条件下的结构与动力学性质。
MS Mesocite的突出特点是使用完全区别于传统介观模拟技术,转而采用力场(Forcefield)方法—比如MS Martini力场—来描述粗粒度之间的相互作用,从而得到体系的结构、和动力学特性,分析函数主要有角度分布,密度分布,径向分布函数,二面角分布,均方根位移等。
分子动力学模拟步骤和意义摘要:一、分子动力学简介二、分子动力学模拟步骤1.准备模型和初始条件2.计算相互作用力3.更新位置和速度4.检查收敛性及输出结果5.重复步骤2-4,直至达到预定模拟时间三、分子动力学模拟意义1.增进对分子结构和性质的理解2.预测分子间相互作用3.优化化学反应条件4.辅助药物设计和材料研究正文:分子动力学是一种计算化学方法,通过模拟分子间的相互作用和运动轨迹,以揭示分子的结构和性质。
这种方法在许多领域具有广泛的应用,如生物化学、材料科学和药物设计等。
分子动力学模拟的主要步骤如下:1.准备模型和初始条件:在进行分子动力学模拟之前,首先需要构建分子模型,包括原子类型、原子间相互作用力等。
同时,为模拟设定初始条件,如温度、压力和分子位置等。
2.计算相互作用力:根据分子模型,利用力学原理(如牛顿第二定律)计算分子间相互作用力。
这些力包括范德华力、氢键、静电相互作用等,对分子的运动和相互作用起关键作用。
3.更新位置和速度:根据相互作用力,对分子的位置和速度进行更新。
通常采用Verlet积分法或Leap-Frog算法等数值方法进行计算。
4.检查收敛性及输出结果:在每次迭代过程中,需要检查模拟的收敛性。
若达到预设的收敛标准,则输出当前时刻的分子结构和性质。
否则,继续进行下一次迭代。
5.重复步骤2-4,直至达到预定模拟时间:分子动力学模拟通常需要进行大量迭代,以获得足够准确的结果。
在达到预定模拟时间后,可得到完整的分子动力学轨迹。
分子动力学模拟在科学研究和实际应用中具有重要意义。
通过模拟,我们可以更好地理解分子的结构和性质,预测分子间的相互作用,从而为实验设计和理论研究提供有力支持。
此外,分子动力学模拟还有助于优化化学反应条件,为药物设计和材料研究提供理论依据。
介观物理的理论和实验研究介观物理是物理学中的一个重要分支,其研究对象是介于微观和宏观之间的物理现象。
在介观物理研究中,理论和实验相辅相成,互相促进,以期深入理解物质结构与性质之间的关系。
一、介观物理的概述介观物理是介于微观和宏观之间的一个领域,其研究的对象通常是尺寸在10纳米到100微米之间的物质。
这个范围是物理学研究中的一个关键阶段,因为在这个尺度范围内,物质的性质既受到微观粒子的影响,又是宏观特征的总和。
二、介观物理的研究方法1. 数学模型:介观物理的研究往往要依靠数学模型。
对于这些模型,需要进行分析和仿真以确定其行为。
2. 实验室技术:介观物理的实验通常需要使用精密仪器和技术。
例如,扫描电子显微镜和隧道电子显微镜可用于观察纳米尺寸的物体。
3. 计算机模拟:介观物理的研究还需要使用计算机模拟,以模拟和预测物质在不同条件下的行为。
三、介观物理的实验研究1. 纳米光学纳米光学是一种利用纳米结构改变光学行为的方法。
这项技术广泛应用于光学传感和纳米光子学方面。
具体应用包括生物传感器和开发新型电子元器件等。
2. 硅纳米加工硅纳米加工是一种主要用于设计微机电系统(MEMS)和纳米器件的加工技术。
其特点是使用光刻技术将图案从光刻掩模转移到硅表面,然后进行干法或湿法刻蚀,最终形成所需形状和结构。
3. 纳米电子学纳米电子学是一种利用纳米尺寸的器件构造电子元器件的技术。
在纳米电子学中,晶体管的尺寸越来越小,从而提高了芯片的集成度,改善了芯片性能。
四、介观物理的理论研究1. 介观物质的物理学介观物质的物理学是一种研究介观尺度物质的物理性质和现象的学科。
其主要研究内容包括介观物质的电学、热学、力学、光学和磁学等方面。
2. 介观体系的非平衡动力学介观体系的非平衡动力学是研究复杂介观体系的运动方式和动力学行为的学科。
此类体系通常有着高度相互耦合的成分,其动态行为充满了干扰和杂乱。
3. 海森堡模型海森堡模型是一种描述量子自旋介质行为的数学模型。
化学反应动力学模拟实验在化学实验中,化学反应动力学模拟实验是一种常见且重要的实验方法。
通过这种实验,我们可以了解化学反应发生的速度规律,探索反应物浓度、温度等因素对反应速率的影响,进而推断出反应机理。
本文将介绍化学反应动力学模拟实验的原理、方法和应用。
首先,化学反应动力学模拟实验是通过控制实验条件,观察反应物质量的变化,从而研究化学反应速率与反应条件的关系的实验方法。
通常,我们可以选择不同的反应物浓度、温度等条件,观察反应速率的变化,进而确定反应的速率方程。
速率方程可以帮助我们理解反应的进行过程,揭示反应物质的具体作用机制。
其次,进行化学反应动力学模拟实验时,需要准备好实验装置和试剂。
一般来说,实验装置包括反应容器、温度控制装置、搅拌器等。
试剂方面,则需要准确称取反应物的质量,并按照一定的比例混合。
在实验过程中,我们可以通过测定反应物质的浓度变化或者生成物质的浓度变化来确定反应的速率。
通过对实验数据的分析,我们可以推导出反应速率方程,从而揭示反应机理。
在实际应用中,化学反应动力学模拟实验具有广泛的应用价值。
首先,它可以帮助我们研究新的化学反应过程,探索新的反应物或反应条件,从而为新材料的研究和开发提供重要参考。
其次,化学反应动力学模拟实验还可以用于环境保护领域。
通过研究化学反应速率与环境因素的关系,我们可以更好地理解环境中化学反应的发生过程,有针对性地采取措施减少污染物的排放。
另外,化学反应动力学模拟实验还可以用于药物化学的研究。
通过研究药物的分子结构与活性之间的关系,我们可以优化药物配方,提高药物的疗效。
总的来说,化学反应动力学模拟实验是一种重要的实验方法,通过这种方法,我们可以深入了解化学反应速率的规律,探究反应机理,为新材料的研究和开发、环境保护和药物设计等领域提供有力支持。
希望通过这篇文章的介绍,读者们对化学反应动力学模拟实验有一个更加清晰的认识,进而加深对化学实验的理解和认识。
介观尺度聚合物动力学模拟一、介绍随着化学、物理、材料学等科学领域的不断发展,液体聚合物在材料、生物、化学等领域广泛应用,但其微观结构和动力学行为的研究仍需发展。
介观尺度聚合物动力学模拟是一种能够研究聚合物微观结构和动力学行为的研究方法,该方法已经得到广泛应用。
本文将介绍介观尺度聚合物动力学模拟的基本原理、研究方法、应用及发展前景。
二、基本原理介观尺度聚合物动力学模拟是一种从宏观到微观的多尺度计算方法,用于模拟聚合物的结构和动力学行为。
该方法通常采用一种统计物理学的方法,将聚合物视为粗粒子或连续介质,通过计算微观单位的运动动力学行为,模拟了宏观过程中的动力学行为。
基于介观尺度聚合物动力学模拟的理论,可以从单分子、分子集聚形成高分子结构,再到整个复杂的聚合物体系进行模拟和预测其在可控条件下调控过程中的行为。
这一过程涉及到多种计算方法,如分子动力学方法、Monte Carlo方法和随机游动方法等。
三、研究方法1. 分子动力学方法分子动力学方法是一种基于牛顿力学的计算方法,用于模拟固体、液体和气体等多种物质的运动。
在聚合物的模拟中,该方法将聚合物视为粗粒子模型,通过计算聚合物的粗粒子在接触后受到的力和运动,模拟整个聚合物体系的运动。
在这个过程中,需要考虑很多因素,如聚合物粗粒子模型的设计,体系温度、体积、压强和模拟时间等等。
2. Monte Carlo方法Monte Carlo方法是一种基于统计学的计算方法,在聚合物模拟中主要用于计算高分子的构象,这种构象指聚合物分子的不同排列方式和空间位置。
在聚合物模拟中,采用基于Metropolis算法的Monte Carlo方法,通过调整聚合物分子的取向和位置,模拟整个高分子结构的构象状态。
3. 随机游动方法随机游动方法是基于随机过程领域的研究方法。
通过分析聚合物分子的随机游动行为,模拟整个聚合物的动力学行为。
在聚合物模拟中,该方法用于研究聚合物的扩散和输运特性,以及聚合物空间结构的特点。
《计算材料学》实验讲义实验八:介观动力学模拟一、前言1、介观模拟简介长期以来,化学家致力于从分子水平研究物质及其变化,而化学工程工作者主要研究物质在宏观体系的行为,介观层次的化学正是联系微观及宏观的桥梁,是从分子到材料的必由之路,同生命过程也有密切的关联。
由于介观模拟能够模拟的空间尺度(纳米到微米)、时间尺度(纳秒到微秒)更大,应用介观模拟方法可以模拟更加复杂的体系,例如:高分子熔体,高分子稀溶液自组装,表面活性剂溶液自组装,磷脂膜等胶体化学,高分子,生物大分子相关的内容。
目前介观模拟的方法很多,例如耗散颗粒动力学模拟方法(dissipative particle dynamics,DPD),它是根据Hoogerbrugge和Koelman提出的一种针对柔性(soft)球模型流体动力学的模拟,并通过引入粒子间的谐振动势,来模拟聚合物的性质;元胞动力学方法(CDS),基于重整化群理论,对时间相关的Ginzburg-Landau方程直接用数值计算的方法在离散空间上进行描述。
其中单个元胞的演化通常用双曲正切函数表示;动态密度泛函方法(DDFT或MesoDyn),应用于高分子体系,建立在粗粒化高斯链模型的基础上,实际上是一个动态的自洽场方法,使用了朗之万方程(Langevin’s equation)来描述体系演化的动力学。
(1)MS-Mesocite简介MS Mesocite是一个基于粗粒度模拟方法的、可以对广泛体系进行模拟研究的分子力学工具集,模拟的对象大小尺寸在纳米到微米尺度范围,相应地,模拟变化的时间范围落在纳秒至微秒区间。
MS Mesocite的模拟对象遍及多种工业领域,比如复合材料、涂料、化妆品以及药物控缓释等,它可以提供流体在平衡态下、在有剪切力存在下以及其它受限制条件下的结构与动力学性质。
MS Mesocite 的突出特点是使用完全区别于传统介观模拟技术,转而采用力场(Forcefield)方法—比如MS Martini力场—来描述粗粒度之间的相互作用,从而得到体系的结构、和动力学特性,分析函数主要有角度分布,密度分布,径向分布函数,二面角分布,均方根位移等。
同时,您还可以使用力场编辑工具对MS Mesocite的力场进行编辑,以获得满足特殊要求的力场,从而拓展了MS Mesocite的应用范围。
应用Mesocite进行动力学模拟时,最主要的是得到精确的力场。
Martini力场,是由Marrink提出的,可以应用于生物分子体系。
Martin力场中包括四种主要的力场类型:极性(polar-P)、非极性(apolar-C)、无极性(nonpolar-N)、带电(charged-Q)。
每种力场类型又分为若干子类型,极性和非极性根据极性高低下分有五种类型(用下坐标1-5表示),无极性和带电的更具氢键结合能力分为四种类型(d-氢键供体,a氢键受体,da-两个都有,o-都没有),这样使得Martini 力场能够更加精确的描述体系性质,应用于更多不同的有机分子体系。
二、实验目的1、了解介观模拟方法及应用领域2、了解Martini力场的3、掌握Mesocite模块的基本操作三、实验内容以下以介观动力学模拟脂质双分子层为例,熟悉Mesocite的基本操作。
1、打开MS,选择created new project,键入CG-bilayer作为工程的名称,点击OK。
本实例是在软件所有参数在默认的情况下进行的,选择Tools-Settings Organizer,选中CG-bilayer,点击Reset。
2、建脂质分子,建模过程要用到Mesostructure toolbar,如在工具栏中没有此建模工具,点击菜单栏中的view-toolbar-mesostructure,调出此建模工具。
(1)点击Bead Types按钮,打开Bead Types 对话框。
点击Properties…按钮,打开Bead Type Properties 对话框,点击Defaults…按钮,设置Mass为72,Radius为2.35,关闭Bead Type Defaults和Bead Type Properties对话框。
在Bead Types对话框中,定义一下珠子类型:C、GL、PO和NC,关闭对话框。
(2)点击Mesomolecule按钮,打开Build Mesomolecule对话框。
定义粗粒化分子,依次选择4个C、1个GL、1个PO、1个GL和4个C,确定不选Randomize order within repeat unit,点击Build按钮。
在Mesomolecule.xsd文件中左击PO珠子,删除Build Mesomolecule对话框中所有的珠子。
选中Add to branch points,点击more…按钮,打开Mesomolecule Branches 对话框。
设置Number of branches to attach为1,关闭对话框。
在Build Mesomolecule对话框中选择1个NC。
点击Build按钮。
(在显示面板中右击,选择Label,打开label对话框,在properties一栏中选择BeadTypeName,点击Apply,可以检测建立的粗粒化分子是不是正确,可以对比下图。
(3)关闭Build Mesomolecule对话框。
在Project Explorer,把Mesomolecule.xsd 文件名改为DPPC.xsd。
我们得到以下粗粒化分子结构:3、更改Martini力场,分配力场,优化脂质分子。
(1)选择Modules -Mesocite - Forcefield Manager或点击Mesocite tools,选择Forcefield Manage,选择MS Martini,点击>>,打开力场文件。
在Project Explorer 中,把文件名改为MSMartiniCIS.off。
(2)打开MSMartiniCIS.off文件,点击Interactions。
在Show interaction下拉选项中选择Angle Bend。
在空白框中,设置Fi 和Fk 到Na 以及Fj 到Qa。
改变Functional Form 为Cosine Harmonic设置TO为120,KO为10.8。
关闭力场文件并保存。
(3)选择Modules | Mesocite | Calculation或点击Mesocite tools选择Calculation;打开Mesocite Calculation对话框,点击Energy,在Forcefield的下拉选项中选择Browse...,在Choose Forcefield对话框中选择MSMartiniCIS.off。
(4)打开DPPC.xsd文件。
按下ALT键,双击任意C类型珠子,选中所有的C 类型珠子。
在Mesocite Calculation对话框中,点击More...打开Mesocite Proparation options对话框,选择C1,点击Assign按钮。
重复此步,为GL、PO、NC分配力场,分配类型如下表所示:BeadTypeName MS Martini Forcefield Type ChargeC C1 0GL Na 0PO Qa -1.0NC Q0 1.0选择PO珠子,在Properties Explorer中,设置Charge为-1,同样把NC设置为1。
(5)在Mesocite Calculation对话框中,点击Setup,改变Task为Geometry Optimization。
点击Run按钮。
得到以下结构:(6)在工具栏中,选择Measure/Change按钮,下拉选项中点击Angel,依次点击左边的C-GL-PO,同样选择右边的PO-GL-C。
此时会显示出两个接近156.50的角度,选在两个角度,在Properties Explorer中,设置Angels为230。
按下ALT键,双击角度,按下Delete。
得到以下分子结构:(7)参照第二步,定义珠子W,用Build Mesomolecule建模工具,建立一个仅包含W的粗粒化分子。
更改文件名为solvent.xsd。
4、建立双分子层结构。
(1)选择Build | Build Mesostructure | Mesostructure Template或点击Mesostructuretoolbar中的Mesostructure Template,打开Build Mesostructure Template对话框。
改变X、YExtents为64,Z Extent为100。
在Filler中,键入solvent。
点击Build按钮。
在Build Mesostructure Template对话框中,改变Former type为Slab。
改变Depth为44.15,Orientation为Along Z。
选中Enable surface packing;在Filler中键入lipid。
点击Add,关闭对话框。
(2)选择Build | Build Mesostructure | Mesostructure或点击Mesostructure toolbar中的Mesostructure ,打开Build Mesostructure对话框。
solvent filler 中的Mesoscale Molecule,选择solvent.xsd。
lipid filler选择优化的DPPC.xsd。
点击Packing,设置Length scale (L)为1,Density为0.00836;不选Randomize conformations。
在Packing中,点击More...按钮,打开Bead Packing Options对话框;双击打开已经优化过的DPPC.xsd。
选择NC,点击Create bead Head set from selection按钮。
按下CTRL + D取消选定,之后按下CTRL键,选择尾部的两个C珠子。
在Bead Packing Options对话框中,改变Bead tag为Tail,点击Create bead Tail set from selection。
关闭对话框。
标记后的DPPC结构如下:(3)双击mesostructure template.msd。
在Build Mesostructure对话框中,点击Build 按钮。
得到下图所示结构:(4)在菜单栏中选择File | Export...,打开Export对话框,在保存类型下拉选项中选择Materials Studio 3D Atomistic Files (*.xsd),点击Options...按钮,打开MSD/MTD Export Options对话框,设置Length scale为1,点击OK。