蛋白分析
- 格式:doc
- 大小:45.50 KB
- 文档页数:10

差异蛋白分析方法差异蛋白分析是一种用于比较不同样本中蛋白质表达差异的方法。
在生物研究中,差异蛋白质的分析对于理解生物学过程的变化、疾病的发生和发展等具有重要意义。
下面将介绍几种常用的差异蛋白质分析方法。
1. 二维凝胶电泳(2-DE):二维凝胶电泳是一种常用的分离和定量蛋白质的方法。
首先,通过等电聚焦将蛋白质在电泳液中按照等电点(pI)分离出不同pI的谱点,然后,使用SDS-PAGE将蛋白质按照分子量分离出不同大小的谱点。
最后,通过染色或质谱分析技术进行蛋白质的可视化和鉴定。
该方法可以同时分析数千种蛋白质,对于差异蛋白质的筛选具有高通量和分辨率高的优势。
2. 差异凝胶电泳(DIGE):差异凝胶电泳是二维凝胶电泳的改进方法。
该方法利用荧光染料(如CyDye)对两组样本中的蛋白质进行荧光标记,然后将两组样本混合后共同进行电泳分离。
在同一个凝胶上,差异蛋白质的表达差异可以通过荧光信号强度的比较来确定。
相比于传统的二维凝胶电泳,DIGE方法的灵敏度更高,并且能够同时分析多个样本,适用于大规模样品分析。
3. 质谱分析:质谱分析是一种常用的蛋白质鉴定和定量方法。
主要有两种方法:基于质谱仪的定性分析和基于同位素标记的定量分析。
前者通过将蛋白质样品利用质谱仪进行断裂和离子化后,通过质谱图谱与数据库对比鉴定其潜在蛋白质;而后者则通过同位素标记技术(如TMT、iTRAQ等)对两组样品中的蛋白质进行标记,然后将标记样品混合后质谱分析,通过同位素峰的比较来定量差异蛋白质的表达水平。
4. 蛋白质芯片技术:蛋白质芯片是一种高通量和高灵敏度的蛋白质分析方法。
它利用固相支持介质上的已知蛋白质或蛋白质片段(如抗体、寡核苷酸)构建芯片,然后将样品中的蛋白质与芯片上的蛋白质发生特异性结合。
通过对芯片上信号的检测和分析,可以确定蛋白质的表达差异。
蛋白质芯片技术具有高通量、高灵敏度和高特异性等特点,可同时检测上千种蛋白质。
5. 高通量测序技术:高通量测序技术也可应用于差异蛋白质的分析。

三种分析蛋白结构域的方法蛋白质是生命体内重要的功能分子,它们通过其特有的三维结构来实现其功能。
蛋白结构域是指蛋白质结构中具有独立功能和收缩性的区域。
分析蛋白结构域的方法对于理解蛋白的功能和机制有重要意义。
以下是三种常用的分析蛋白结构域的方法。
第一种方法是比对分析。
比对分析是通过比对已知结构域的蛋白质序列和结构与待研究蛋白质序列和结构进行对比,以此来鉴定待研究蛋白质中的结构域。
比对分析常用的工具有BLAST和HMMER等。
BLAST(基本局部序列比对工具)通过比对两个蛋白序列的共同片段来确定相似性,可以帮助确定蛋白质的结构域。
HMMER(隐含马尔可夫模型比对工具)则建立了一个隐含马尔可夫模型,将待研究的蛋白质序列与已知结构域的蛋白质序列进行比对,以此来确定结构域。
第二种方法是结构预测。
结构预测是通过计算机程序对蛋白质序列进行建模,以预测其三维结构。
常见的结构预测方法有基于比对的序列相似性建模、基于物理力学的方法和基于机器学习的方法等。
基于比对的序列相似性建模方法通过比对已知结构域的蛋白质序列与待研究蛋白质序列来构建模型,以此来预测待研究蛋白质的结构域。
基于物理力学的方法则基于分子力学和物理化学原理,通过计算机模拟来推测蛋白质的结构。
基于机器学习的方法则使用已知结构域的蛋白质数据来训练算法,以此来预测待研究蛋白质的结构域。
第三种方法是功能簇分析。
功能簇分析是通过聚类算法来将蛋白质分为不同的簇,以确定其中的结构域。
常见的聚类算法有层次聚类、基于密度的聚类和K均值聚类等。
层次聚类是将样本逐步合并成不同的簇,直到达到预定的停止条件。
基于密度的聚类则是根据样本的密度将其分为不同的簇。
K均值聚类是将样本分为K个不同的簇,使得簇内的样本之间的差异最小化。
通过功能簇分析可以鉴定出具有相似功能的蛋白质结构域。
综上所述,比对分析、结构预测和功能簇分析是常用的分析蛋白结构域的方法。
这些方法能够帮助鉴定蛋白质中的结构域,进而理解其功能和机制。

蛋白检测方法蛋白是生物体内非常重要的一类生物大分子,它参与了生物体内的许多重要生命活动,包括细胞信号传导、酶活性调控、结构支持等。
因此,对蛋白进行检测和分析对于生命科学研究具有重要意义。
本文将介绍几种常见的蛋白检测方法,希望能够为科研工作者提供一些参考和帮助。
1. SDS-PAGE凝胶电泳。
SDS-PAGE凝胶电泳是一种常用的蛋白质分离和检测方法。
它通过聚丙烯酰胺凝胶电泳,根据蛋白质的分子量进行分离,然后通过染色或Western blotting等方法进行检测。
这种方法操作简单,成本低廉,可以快速得到蛋白质的分子量信息。
2. 免疫沉淀。
免疫沉淀是利用抗体对特定蛋白的特异性结合来进行蛋白的富集和检测。
通过将样品与特异性抗体结合,然后利用蛋白A/G琼脂糖或其他方法将抗体-蛋白复合物沉淀下来,最后进行Western blotting或质谱分析等方法进行检测。
3. 酶联免疫吸附实验(ELISA)。
ELISA是一种高灵敏度、高特异性的蛋白检测方法。
它利用酶标记的抗体对特定蛋白进行捕获和检测,通过底物的酶促反应产生可定量的信号。
ELISA方法广泛应用于临床诊断、生物医药研究等领域。
4. 质谱分析。
质谱分析是一种高分辨率、高灵敏度的蛋白质检测方法。
通过质谱仪对蛋白质进行分析,可以得到蛋白质的分子量、氨基酸序列、修饰信息等。
质谱分析方法适用于复杂混合物的蛋白质检测和鉴定。
5. 蛋白质芯片技术。
蛋白质芯片技术是一种高通量、高平行性的蛋白质检测方法。
它通过将大量的蛋白质捕获到芯片表面,然后利用荧光标记、质谱分析等方法进行检测。
蛋白质芯片技术在蛋白质相互作用、信号通路等研究中具有重要应用价值。
总结。
蛋白检测方法的选择应根据具体的实验目的、样品特性和实验条件来确定。
不同的方法各有优缺点,科研工作者应根据实际需求选择合适的方法进行蛋白检测和分析。
希望本文介绍的蛋白检测方法对您有所帮助,祝您的科研工作顺利!。
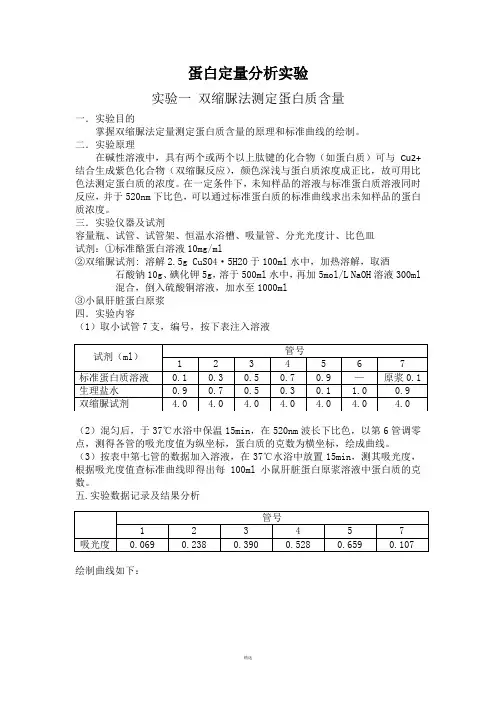
蛋白定量分析实验实验一双缩脲法测定蛋白质含量一.实验目的掌握双缩脲法定量测定蛋白质含量的原理和标准曲线的绘制。
二.实验原理在碱性溶液中,具有两个或两个以上肽键的化合物(如蛋白质)可与Cu2+结合生成紫色化合物(双缩脲反应),颜色深浅与蛋白质浓度成正比,故可用比色法测定蛋白质的浓度。
在一定条件下,未知样品的溶液与标准蛋白质溶液同时反应,并于520nm下比色,可以通过标准蛋白质的标准曲线求出未知样品的蛋白质浓度。
三.实验仪器及试剂容量瓶、试管、试管架、恒温水浴槽、吸量管、分光光度计、比色皿试剂:①标准酪蛋白溶液10mg/ml②双缩脲试剂: 溶解2.5g CuSO4·5H2O于100ml水中,加热溶解,取酒石酸钠10g、碘化钾5g,溶于500ml水中,再加5mol/L NaOH溶液300ml混合,倒入硫酸铜溶液,加水至1000ml③小鼠肝脏蛋白原浆四.实验内容(1)取小试管7支,编号,按下表注入溶液(2)混匀后,于37℃水浴中保温15min,在520nm波长下比色,以第6管调零点,测得各管的吸光度值为纵坐标,蛋白质的克数为横坐标,绘成曲线。
(3)按表中第七管的数据加入溶液,在37℃水浴中放置15min,测其吸光度,根据吸光度值查标准曲线即得出每100ml小鼠肝脏蛋白原浆溶液中蛋白质的克数。
五.实验数据记录及结果分析绘制曲线如下:由图可知,当吸光度为0.107时,相对应的蛋白质克数为1.38 mg,则100ml 小鼠肝脏蛋白原浆液中蛋白质克数为1.38 g。
实验二考马斯亮蓝法测定蛋白质含量一.实验目的掌握考马斯亮蓝法定量测定蛋白质含量的原理和方法;熟悉紫外分光光度计的使用。
二.实验原理考马斯亮蓝在游离状态下呈红色,当它与蛋白质结合后变为蓝色,在一定蛋白质浓度范围内,结合物在595 nm波长下有最大吸收峰,测其光的吸收量即可得结合蛋白质的量。
三.实验仪器及试剂仪器:分光光度计,试管,吸量管,比色皿试剂:①考马斯亮蓝试剂: 考马斯亮蓝G-250 100mg溶于50 ml 95%乙醇中,加入100ml 85%磷酸,用蒸馏水稀释至1000 ml。

一、实验目的1. 掌握蛋白质定量分析的基本原理和方法。
2. 学习使用紫外分光光度计进行蛋白质定量。
3. 了解不同蛋白质定量方法的优缺点,为后续实验提供参考。
二、实验原理蛋白质定量分析是生物化学实验中常用的技术之一,主要方法有直接紫外吸收法、Bradford法和Lorry法等。
本实验主要采用直接紫外吸收法和Bradford法进行蛋白质定量。
1. 直接紫外吸收法:蛋白质分子中含有共轭双键的酪氨酸和色氨酸,在280nm波长附近有特征吸收峰。
蛋白质溶液的光密度(OD)与蛋白质浓度呈正比,因此可以通过测定OD值来计算蛋白质浓度。
2. Bradford法:蛋白质与染料考马斯亮蓝G-250结合,在一定线性范围内,反应液595nm处吸光度的变化量与蛋白质量成正比,从而实现蛋白质定量。
三、实验材料与仪器1. 实验材料:蛋白质样品、标准蛋白质溶液、考马斯亮蓝G-250染料、双蒸水等。
2. 仪器:紫外分光光度计、电子天平、移液器、试管等。
四、实验步骤1. 标准曲线绘制(1)取6支试管,分别加入0、0.1、0.2、0.4、0.6、0.8、1.0ml标准蛋白质溶液,用双蒸水补至1.0ml。
(2)分别加入5ml考马斯亮蓝G-250染料,混匀。
(3)在595nm波长下测定各管吸光度值。
(4)以标准蛋白质浓度为横坐标,吸光度值为纵坐标,绘制标准曲线。
2. 蛋白质定量(1)取6支试管,分别加入0、0.1、0.2、0.4、0.6、0.8、1.0ml蛋白质样品,用双蒸水补至1.0ml。
(2)分别加入5ml考马斯亮蓝G-250染料,混匀。
(3)在595nm波长下测定各管吸光度值。
(4)根据标准曲线,计算蛋白质浓度。
五、实验结果与分析1. 标准曲线绘制绘制标准曲线,得到线性方程:y = 0.004x + 0.021,相关系数R² = 0.997。
2. 蛋白质定量根据标准曲线,计算蛋白质样品浓度,结果如下:样品1:0.5mg/ml样品2:1.0mg/ml样品3:1.5mg/ml样品4:2.0mg/ml样品5:2.5mg/ml样品6:3.0mg/ml六、讨论与心得1. 实验过程中,应注意操作规范,避免蛋白质样品污染。

蛋白质的定量和定性分析方法蛋白质是生物体内最重要的功能分子之一,对于研究生物体的结构和功能具有重要意义。
为了准确地了解蛋白质的含量和性质,在科学研究和实际应用中,我们需要使用定量和定性分析的方法来研究蛋白质。
一、定量分析方法1. 低里德伯法(Lowry method)低里德伯法是一种经典而广泛应用的蛋白质定量方法。
该方法利用蛋白质与碱式铜络合物在碱性条件下反应生成蓝色产物,通过比色法测定溶液的吸光度来计算蛋白质含量。
这是一种灵敏且相对简单的方法,适用于大多数蛋白质样品的定量分析。
2. 比色法(Colorimetric assay)比色法是一种常用的蛋白质定量方法,通过蛋白质与染料的结合来测定蛋白质浓度。
常用的染料有布拉德福蓝(Bradford)、库吉铃蓝(Coomassie Brilliant Blue)、BCA法(Bicinchoninic Acid assay)等。
这些染料与蛋白质结合后形成一种复色物,通过比色法测定溶液的吸光度可以定量分析蛋白质。
比色法具有操作简便、灵敏度高等特点,被广泛应用于蛋白质定量领域。
3. 分子标记法(Molecular tagging method)分子标记法是一种新兴的蛋白质定量方法,利用特定的分子标记物(如荧光染料、放射性示踪剂等)标记蛋白质,然后通过测定标记物的荧光强度或放射性信号来计算蛋白质浓度。
分子标记法具有高灵敏度、高特异性等优点,适用于微量蛋白质的定量测定。
二、定性分析方法1. SDS-PAGE(Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis)SDS-PAGE是一种常用的蛋白质定性分析方法,通过电泳将蛋白质在聚丙烯酰胺凝胶中分离出来。
在电泳过程中,蛋白质在SDS(十二烷基硫酸钠)的作用下具有相同的电荷密度,只受到大小的限制而移动。
蛋白质在凝胶中的分离程度取决于其分子量大小,可以通过对比标准品的迁移距离来估计样品中蛋白质的相对分子量。

蛋白鉴定分析报告1. 引言蛋白质是生物体内不可或缺的重要分子,其在细胞内扮演着各种生物学功能的角色。
蛋白质的结构和功能对于了解生物体的生命过程以及疾病的发生和治疗具有重要意义。
本报告旨在对某一蛋白质进行鉴定分析,并提供相关的数据和结果。
2. 实验方法在蛋白质鉴定分析中,我们采用了以下的实验方法:•样品准备:收集蛋白质样品,并进行必要的处理和预处理工作,如蛋白提取和纯化。
•SDS-PAGE:采用聚丙烯酰胺凝胶电泳(Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis, SDS-PAGE)技术,对蛋白质样品进行分离。
•蛋白质染色:对SDS-PAGE分离后的蛋白质进行染色,以观察蛋白质的迁移位置和相对分子质量。
•质谱分析:采用质谱技术,如质谱仪和液相色谱质谱联用技术(Liquid Chromatography-Mass Spectrometry, LC-MS),对蛋白质进行分析和鉴定。
•数据处理和分析:对实验得到的数据进行处理和分析,包括蛋白质质量谱图的解析和蛋白质鉴定。
3. 结果与讨论3.1 SDS-PAGE分析结果在SDS-PAGE实验中,我们观察到蛋白质样品在凝胶上呈现出明显的迁移带。
根据迁移带的位置和相对分子质量,我们可以初步判断蛋白质的大小和纯度。
3.2 质谱分析结果在质谱分析中,我们获得了蛋白质的质谱图和相关的质谱数据。
通过对质谱图的解析和蛋白质鉴定数据库的比对,我们得到了蛋白质的鉴定结果。
根据质谱数据分析,我们确定了蛋白质的氨基酸组成、序列以及可能的修饰模式。
此外,我们还对蛋白质的结构和功能进行了初步的预测和分析。
3.3 结果讨论根据实验结果和分析,我们得出了以下的结论:•蛋白质的分子质量:根据SDS-PAGE实验结果和质谱分析数据,我们可以确定蛋白质的相对分子质量范围。
•蛋白质的鉴定结果:通过质谱分析,我们成功鉴定了蛋白质的氨基酸组成和序列,并预测了其可能的结构和功能。

蛋白质结构解析方法
1. X 射线衍射法,这就像是给蛋白质拍一张超级清晰的照片呀!你想想看,通过X 射线的照射,我们就能看清蛋白质的三维结构,那得多厉害啊,就像我们用高清相机拍出美丽的风景一样。
2. 核磁共振法,哇塞,这就仿佛是在和蛋白质进行一场深入的对话呢!它能告诉我们一个个原子的位置信息,是不是很神奇,就好比我们和好朋友聊天,能了解到对方内心的小秘密呀。
3. 冷冻电镜法,哎呀呀,这简直就是给蛋白质来个大特写嘛!能让我们看到极其细微的结构,这多让人兴奋啊,就像近距离观察一朵盛开的鲜花的每一个花瓣细节一样。
4. 质谱分析法,嘿,这不就是个厉害的检测神器嘛!可以分析蛋白质的组成成分呢,就像一个超级侦探能找出各种小线索一样哩。
5. 荧光光谱法,哇哦,这就好像是给蛋白质打上了独特的光芒呀!让我们能更好地了解它,这感觉是不是超酷的,就跟舞台上的聚光灯照亮演员一样。
6. 圆二色性光谱法,嘿嘿,这就如同给蛋白质穿上了一件能显示特征的衣服呀!通过它我们能知道蛋白质的结构特征呢,是不是很奇妙呀,就像我们根据一个人的穿着打扮来判断他的风格一样。
7. 氢氘交换法,呀,这相当于在研究蛋白质时给它来个特别的标记呢!能帮助我们深入探究其结构变化,这多有意思呀,就像给一个物品做个独特的记号一样。
8. 等温滴定量热法,哇,这可是能测量蛋白质相互作用的神奇方法呢!能让我们知道它们之间的关系,是不是超级棒,就像我们了解人与人之间是如何互动交往的一样。
我觉得这些蛋白质结构解析方法都太了不起啦,每一种都像是一把开启蛋白质奥秘的钥匙,让我们能不断深入了解这个神奇的微观世界!。

生物化学实验中的蛋白质分析技术蛋白质分析技术在生物化学实验中的应用在生物化学实验中,蛋白质分析技术是一项十分重要的技术。
蛋白质是生物体中最基本的分子组成部分之一,对于研究生物体的生化过程和功能具有重要意义。
本文将介绍几种常用的蛋白质分析技术,包括SDS-PAGE、Western Blot、质谱分析和免疫沉淀等。
重点讲述这些技术的原理、操作步骤以及其在生物化学实验中的应用。
一、SDS-PAGE技术SDS-PAGE(Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis)是一种常用的蛋白质分析技术,通过电泳的方式将蛋白质样品分离成不同的电泳带来研究其分子量和组成。
1. 原理:SDS-PAGE利用带负电荷的SDS使蛋白质样品具有净电荷,根据蛋白质分子量的不同,通过电泳的方式将蛋白质分离到聚丙烯酰胺凝胶中,然后用染色方法可视化蛋白质电泳带。
2. 操作步骤:制备凝胶、样品处理、电泳、染色等。
3. 应用:常用于估计蛋白质的分子量、纯度和相对表达水平等。
二、Western Blot技术Western Blot是一种用于检测特定蛋白质的技术,常用于研究蛋白质的表达、定位和相互作用等。
1. 原理:Western Blot主要由蛋白质电泳分离、转膜、蛋白质与抗体的特异性结合以及信号检测等步骤组成。
2. 操作步骤:SDS-PAGE分离蛋白质、转膜、抗体孵育、信号检测等。
3. 应用:常用于检测特定蛋白质在不同样品中的表达差异、研究蛋白质的翻译后修饰等。
三、质谱分析技术质谱分析技术是一种可以确定蛋白质分子量和氨基酸序列的方法,广泛应用于蛋白质鉴定和结构研究等领域。
1. 原理:质谱分析技术常用的方法有质谱图谱分析和串联质谱分析。
2. 操作步骤:样品制备、质谱分析、数据解析等。
3. 应用:常用于蛋白质的鉴定、研究蛋白质的翻译后修饰、蛋白质定量等。
四、免疫沉淀技术免疫沉淀技术是一种通过特异性抗体与特定蛋白质结合,进而将目标蛋白质从混合物中分离出来的方法,常用于研究蛋白质相互作用以及功能等。

生物信息学中的蛋白质分析技术蛋白质是生物体中不可或缺的重要分子,其功能包括酶催化、信号传递、结构支持等多种生命活动。
蛋白质分析是生物信息学研究中的重要领域之一,目的是从生物样品中获取有关蛋白质的信息。
这项技术不仅可以揭示蛋白质的结构和功能,还可以为医学诊断和药物研发提供重要的参考。
一、蛋白质分析的基本流程蛋白质分析的基本流程包括蛋白质提取、分离纯化、分析鉴定等几个步骤。
蛋白质提取是将目标蛋白从生物样品中提取出来,一般采用机械破碎、化学分解、超声波等方法。
分离纯化是将目标蛋白与其他蛋白分离开来,可以采用电泳、层析、过滤等方法。
分析鉴定则是对分离得到的蛋白进行化学、物理和生物学的分析,如质谱分析、核酸测序、免疫学检测等方法。
二、质谱分析技术的应用质谱分析是一种可以同时检测多种蛋白质组成和结构的方法,其技术基础是将蛋白质分离并进行离子化后进行质量分析。
这种方法被广泛地应用于蛋白质组学和蛋白质互作等领域。
在蛋白质组学中,将样品中的所有蛋白质分离并进行质谱分析,可以获得大量的信息,如蛋白质的数量、种类、分布和修饰状态等。
质谱分析技术的应用还包括蛋白质互作的研究。
蛋白质互作通常是指两个或多个蛋白质之间的相互作用,这在生物活动中非常重要。
质谱分析可以用来鉴定已知的蛋白质互作或发现新的蛋白质互作,这对于深入理解生物活动机理具有重要意义。
三、结构生物学的应用结构生物学是研究蛋白质三维结构的一种技术,其目的是探究蛋白质结构与功能之间的关系。
现有的结构生物学技术主要包括X射线晶体学、核磁共振和电子显微镜。
通过这些技术,可以确定单个蛋白质的原子结构,也可以确定蛋白质的超分子结构,如蛋白质-DNA复合物和蛋白质-蛋白质复合物等。
在药物研发方面,结构生物学的应用也非常广泛。
通过了解蛋白质的结构,可以设计出针对特定靶标的药物,并对药物与靶标之间的相互作用进行优化和改良。
四、生物信息学的应用生物信息学是将计算机和数学等方法应用于生物学研究的一种学科。

蛋白质纯度分析方法
蛋白质纯度分析方法有多种,常用的包括:
1. SDS-PAGE(聚丙烯酰胺凝胶电泳): 这是一种常用的蛋白质纯度分析方法。
在这种方法中,蛋白质样品被加入到聚丙烯酰胺凝胶中,经过电泳分离,根据分子量的大小在凝胶上形成多个蛋白带,通过染色或蛋白质标记物与凝胶上的蛋白质结合后观察,可以确定蛋白质的纯度和相对分子量。
2. 高效液相色谱(HPLC): 这是一种利用溶液相流动将混合物中的成分进行分离和纯化的方法。
蛋白质在HPLC柱中按照其理化性质如大小、极性和亲水性等进行分离,可以通过检测紫外光吸收、荧光或质谱等来确定蛋白质的纯度。
3. 硫酸铵沉淀: 这是一种通过加入一定浓度的硫酸铵使蛋白质发生沉淀而进行
纯化的方法。
纯化后的蛋白质样品可以通过比色法或质谱分析等方法来确定纯度。
4. 分子筛层析: 这是一种通过蛋白质分子大小的差异来进行分离和纯化的方法。
蛋白质混合物经过分子筛层析柱时,分子较小的蛋白质能够进入分子筛的孔隙中,而分子较大的蛋白质则被排除在外,从而实现对蛋白质纯化的目的。
5. 亲和层析: 这是一种利用蛋白质与某种亲和固定相之间的特定相互作用进行
分离和纯化的方法。
亲和相可以是具有特定配体的固定相,如金属离子、抗体、
亲和标签等,蛋白质与亲和相结合后,其他非特异性结合的蛋白质被洗去,再用特定条件将目标蛋白质洗脱,从而实现对蛋白质的纯化。
蛋白质定量分析方法蛋白质定量是生物学和生物化学实验中常用的一项分析方法,用于测量样品中的蛋白质浓度。
正确的蛋白质定量对于实验结果的准确性和可重复性至关重要。
以下将介绍几种常用的蛋白质定量方法。
1. 布拉德福法:布拉德福法是一种经典的蛋白质定量方法。
它基于蛋白质与染料结合产生颜色变化的原理。
该方法使用的布拉德福染料与蛋白质中的酪氨酸和苯丙氨酸反应,生成一个特定的吸收波长下的蓝色色素。
通过比较标准曲线上已知浓度的蛋白质溶液和待测样品的吸光度,可以推算出待测样品的蛋白质浓度。
2. 低里德伯法:低里德伯法是一种基于腐蚀性或氧化性试剂使蛋白质产生特殊反应而定量的方法。
其中最常用的是使用硫酸硫酸氨基酸反应,将蛋白质转化为酸解蛋白质,然后测定在特定条件下副产物的吸收光度。
通过比较标准曲线上已知浓度的蛋白质标准品和待测样品的吸光度,可以计算出待测样品中蛋白质的浓度。
3. BCA法:BCA法是一种受欢迎的蛋白质定量方法,也是一种基于蛋白质染料结合并产生颜色变化的原理。
BCA(Bicinchoninic Acid)试剂与蛋白质中的蛋白质肽键和酪氨酸、苯丙氨酸等残基反应生成紫色络合物。
通过比较标准曲线上已知浓度的蛋白质溶液和待测样品的吸光度,可以计算出待测样品中蛋白质的浓度。
4. 比色法:比色法是一种使用标准蛋白质溶液与待测样品比较颜色的方法。
其中最常用的是使用深蓝色染料康氏试剂。
标准蛋白质溶液与康氏试剂反应生成一个蓝色络合物,该络合物的吸光度与蛋白质的浓度成正比。
通过比较标准曲线上已知浓度的蛋白质溶液和待测样品的吸光度,可以计算出待测样品中蛋白质的浓度。
除了以上几种常用的蛋白质定量方法之外,还有其他一些描述复杂的方法,如ELISA、酶联免疫吸附试验等。
这些方法在定量蛋白质方面有其独特的优势和适用范围。
总结起来,蛋白质定量是实验室中广泛应用的一种分析方法。
根据不同的研究目的和实验条件,可以选择适用的方法来准确测量样品中蛋白质的浓度。
蛋白质分析方法1、微量凯氏(Kjeldahl)定氮法样品与浓硫酸共热。
含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。
经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。
若以甘氨酸为例,其反应式如下:NH2CH2COOH+3H2SO4——2CO2+3SO2+4H2O+NH3 (1)2NH3+H2SO4——(NH4)2SO4 (2)(NH4)2SO4+2NaOH——2H2O+Na2SO4+2NH3 (3)反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。
为了加速消化,可以加入CuSO4作催化剂,K2SO4以提高溶液的沸点。
收集氨可用硼酸溶液,滴定则用强酸。
实验和计算方法这里从略。
计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白氮即得。
如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。
评价:总氮-非蛋白氮=蛋白质氮——>蛋白质含量灵敏度低,误差大,耗时长。
2、双缩脲法(Biuret法)(一)实验原理双缩脲(NH3CONHCONH3)是两个分子脲经180℃左右加热,放出一个分子氨后得到的产物。
在强碱性溶液中,双缩脲与CuSO4形成紫色络合物,称为双缩脲反应。
凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。
紫色络合物颜色的深浅与蛋白质浓度成正比,而与蛋白质分子量及氨基酸成分无关,故可用来测定蛋白质含量。
测定范围为1-10mg蛋白质。
干扰这一测定的物质主要有:硫酸铵、Tris 缓冲液和某些氨基酸等。
此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。
主要的缺点是灵敏度差。
因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。
(二)试剂与器材1. 试剂:(1)标准蛋白质溶液:用标准的结晶牛血清清蛋白(BSA)或标准酪蛋白,配制成10mg/ml 的标准蛋白溶液,可用BSA浓度1mg/ml的A280为0.66来校正其纯度。
蛋白质鉴定分析报告1. 简介蛋白质是生物体中一类非常重要的生物大分子,它在生命活动中发挥着关键作用。
蛋白质鉴定分析是一项广泛应用于生物学研究和临床诊断的技术,通过对蛋白质的结构、功能和相互作用进行分析,可以揭示其在生命过程中的具体功能和调控机制。
2. 样品准备在进行蛋白质鉴定分析之前,首先需要准备好样品。
样品可以是从生物体中提取的组织、细胞或体液,也可以是已经纯化的蛋白质。
对于复杂的生物样品,通常需要进行蛋白质提取和富集,以去除干扰物质并增加蛋白质的浓度。
在样品准备过程中,要注意避免蛋白质的降解和污染,以保证后续分析的准确性和可靠性。
3. 蛋白质分离与纯化蛋白质的复杂性和多样性使得其分离与纯化成为鉴定分析的重要步骤。
常用的蛋白质分离技术包括凝胶电泳、液相色谱和电泳等。
其中,凝胶电泳是一种常用的分离方法,可以根据蛋白质的分子量、电荷和等电点进行分离。
液相色谱则可以根据蛋白质的亲和性、分配系数和分子尺寸进行分离。
通过这些分离方法,可以获得纯度较高的蛋白质样品,为后续鉴定分析提供可靠的基础。
4. 质谱分析质谱分析是蛋白质鉴定分析的关键步骤之一。
质谱技术可以通过测量蛋白质样品中离子的质荷比来确定其分子量,并通过与数据库中已知蛋白质序列进行比对,从而确定样品中蛋白质的身份。
常用的质谱技术包括质谱仪、蛋白质鉴定软件和数据库等。
质谱分析可以提供蛋白质的分子量、序列以及修饰信息,为进一步的功能和相互作用分析提供基础。
5. 功能和相互作用分析蛋白质的功能和相互作用是其在生命过程中发挥作用的关键。
功能分析可以通过生物学实验和生物信息学方法来揭示,例如酶活性测定、结构预测和功能注释等。
相互作用分析则可以通过蛋白质的亲和性、结合能力和结构构象等特性来研究。
这些分析方法可以揭示蛋白质在细胞信号传导、代谢调节和疾病发生等过程中的具体作用机制。
6. 结论与展望蛋白质鉴定分析是一项复杂而有挑战性的工作,但其在生物学研究和临床诊断中具有重要的应用价值。
蛋白质定量分析方法蛋白质定量是蛋白质研究中非常重要的一项工作,它用于确定样品中蛋白质的浓度。
在蛋白质研究中,常常需要确定样品中蛋白质的浓度,以便进行后续的实验和数据分析。
当前常用的方法包括光谱法、比色法、生物学活性测定法和质谱法等。
下面将对这些方法进行详细阐述。
光谱法是一种常用的蛋白质定量方法,它是通过测量蛋白质溶液在特定波长下的吸光度来确定蛋白质的浓度。
在这种方法中,常用的波长是280nm,因为大部分蛋白质在这个波长下有较高的吸光度。
通过测量蛋白质溶液的吸光度值,可以使用比色法或者标准曲线法来计算蛋白质的浓度。
光谱法的优点是操作简单,不需要复杂的设备和试剂,而且可以同时测定多种蛋白质。
但是,光谱法对于一些特殊蛋白质和杂质的检测有一定的局限性。
比色法是蛋白质定量中常用的一种方法,它是通过比较样品和标准溶液的颜色差异来确定蛋白质的浓度。
比色法的原理是,蛋白质与某些特定试剂反应后,可以形成有颜色的复合物。
一般来说,蛋白质和某种染料或金属离子反应会产生特定的吸光峰,通过测量这个吸光峰的强度,可以确定蛋白质的浓度。
比色法的优点是快速、准确,适用范围广,且可以同时测定多种样品。
然而,比色法也存在一些问题,比如可能受到样品中其他物质的干扰,以及对试剂的选择和操作有一定的要求。
生物活性测定法是一种基于蛋白质的生物学活性特性来确定蛋白质浓度的方法。
在这种方法中,通过测定蛋白质的生物学活性,如酶活性、生物修复能力等,来推断蛋白质的浓度。
生物活性测定法在一些特定情况下非常有用,比如检测蛋白质的功能状态或者酶的活性。
但是,生物活性测定法也存在一些问题,如操作复杂、结果受到其他因素的干扰等。
质谱法是一种高灵敏度的蛋白质定量方法,它是通过测定样品中蛋白质产生的质荷比来确定蛋白质的浓度。
质谱法可以使用质谱仪来进行分析,通过电离和分离蛋白质,然后测定质荷比,计算样品中蛋白质的浓度。
质谱法具有高灵敏度、高分辨率和高通量的优点,能够检测到非常低浓度的蛋白质。
蛋白质结构分析方法蛋白质是生物体中重要的功能分子,其结构对其功能起着至关重要的作用。
因此,了解蛋白质的结构对于深入理解其功能和参与药物设计、生物工程等领域的研究具有重要意义。
蛋白质的结构包括其空间构型、二级结构和三级结构等层次。
下面将介绍一些常见的蛋白质结构分析方法。
1.X射线晶体学:这是分析蛋白质结构最常用且最直接的方法。
通过蛋白质晶体与X射线的相互作用,得到蛋白质的高分辨率结构。
这种方法的优势是可以提供非常精确的原子级别的结构信息,但需要得到高质量的蛋白质晶体。
2.光学方法:包括圆二色光谱、拉曼光谱等。
圆二色光谱是根据蛋白质结构中的手性部分对偏振光的旋转度进行测量,从而得到蛋白质的二级结构信息。
拉曼光谱则是通过测量蛋白质结构中的振动模式,来揭示蛋白质的分子间相互作用和结构变化。
3.核磁共振(NMR):这是一种无需蛋白质晶体的方法,可以在溶液中研究蛋白质的结构。
通过测量蛋白质中核磁共振现象的信号,可以得到蛋白质的二级和三级结构信息。
4.电子显微镜(EM):这种方法可以提供蛋白质的结构信息,尤其适用于大型复合物的研究。
通过显微镜观察和图像处理技术,可以获得近原子级别的结构信息。
5.质谱(MS)方法:这种方法可以用于蛋白质的质量鉴定和结构分析。
质谱技术通常用于测量蛋白质的分子量、氨基酸序列和翻译后修饰等信息。
除了上述方法外,还有许多辅助分析方法可以结合使用来解析蛋白质的结构。
例如,计算化学方法可以通过建模、模拟等手段预测蛋白质的结构。
此外,还可以利用蛋白质的化学性质和酶切等策略进行结构解析。
总之,蛋白质结构分析方法多种多样,各有其优势和应用范围。
通过这些方法的结合应用,我们可以更加深入地了解蛋白质的结构和功能,从而为药物设计、生物工程等领域的研究提供基础和指导。
蛋白定量分析实验报告简介蛋白质是生物体中最重要的分子之一,它们扮演着许多生物功能的关键角色。
蛋白质定量分析是研究蛋白质含量和表达水平的重要手段。
本实验报告旨在介绍一种常用的蛋白定量分析方法。
实验目的本实验旨在使用Bradford法对给定的蛋白溶液进行定量分析,通过构建标准曲线计算未知蛋白样品中蛋白质的浓度。
实验步骤1. 制备标准曲线1.准备一系列已知浓度的蛋白溶液,浓度范围从0到1.0 mg/mL。
2.分别取0.2 mL标准蛋白溶液并加入1.8 mL Bradford试剂。
将混合液在室温下孵育5分钟。
3.使用分光光度计在595 nm波长下测量吸光度,并记录吸光度值。
2. 测定未知样品的蛋白质浓度1.取待测蛋白样品0.2 mL,并加入1.8 mL Bradford试剂。
将混合液在室温下孵育5分钟。
2.使用分光光度计在595 nm波长下测量吸光度,并记录吸光度值。
3.使用标准曲线计算未知样品的蛋白质浓度。
3. 数据处理1.绘制标准曲线,横轴表示已知蛋白质浓度,纵轴表示对应的吸光度值。
2.使用线性回归等方法拟合标准曲线,得到拟合方程。
3.根据未知样品的吸光度值和拟合方程,计算未知样品的蛋白质浓度。
结果与讨论我们使用Bradford法对一系列已知浓度的蛋白溶液进行了吸光度测量,并绘制了标准曲线。
通过拟合标准曲线,我们得到了一个线性方程:浓度(mg/mL)= 0.73 × 吸光度 - 0.02。
然后,我们对一个未知蛋白样品进行了吸光度测量,并使用拟合方程计算出样品中蛋白质的浓度为0.62 mg/mL。
通过本实验,我们成功地确定了未知样品中蛋白质的浓度。
这种蛋白定量分析方法简单、快速且可靠,可广泛应用于生物化学和生命科学领域。
结论本实验使用Bradford法对蛋白样品进行定量分析。
通过制备标准曲线和测定未知样品的吸光度值,我们成功地确定了未知样品中蛋白质的浓度为0.62 mg/mL。
这种方法简单易行且结果可靠,是一种常用的蛋白定量分析方法。
Gi:40644130 allene oxide cyclase 【Niootiana tabacum】丙二烯氧化环化酶(allene oxide cyclase, AOC)Gi:140083805 cytosolic class II small heatshock protein HSP17.5 【Rosa hybrid cultivar】Gi:289487897 Lasoorbate peroxidasa 【Bruguiera gymnorhiza】比如是核糖体16S, 18S,或ITS等DNA序列,一般在Blast n 中搜索,到底是用megablast,discontiguous megablast还是blastn要根据你的序列与数据库序列的相似性,一般首先用blastn,它对相似性要求较低,可发现大量相似序列,如果进一步要求,再选择megablast 等。
但是注意blastn搜索数据库对核酸序列相似性要求较高,如果序列保守性不高,比如新的RNA病毒的基因组序列,可能很难得到结果,这时需要用blastp 或Blast x等。
如果是编码蛋白的基因序列,可先将其翻译成蛋白(注意,一条序列理论上有六种编码可能)然后分别去blastp搜索蛋白数据库,当然,你也可直接将其在Blast x中搜索,Blast x会自动将六种编码可能分别翻译后搜索蛋白数据库。
Blastp/PSI-Blast/PHI-BLAST都是蛋白序列与蛋白序列之间的Blast比对。
1,Blastp: 标准的蛋白序列与蛋白序列之间的比对Standard protein BLAST is designed for protein searches.Blastp用于确定查询的氨基酸序列在蛋白数据库中找到相似的序列。
跟其它的Blast程序一样,目的是要找到相似的区域。
2,PSI-BLAST : 敏感度更高的蛋白序列与蛋白序列之间的比对PSI-BLAST is designed for more sensitive protein-protein similarity searches.Position-Specific Iterated (PSI)-BLAST,是一种更加高灵敏的Blastp程序,对于发现远亲物种的相似蛋白或某个蛋白家族的新成员非常有效。
当你使用标准的Blastp比对失败时,或比对的结果仅仅是一些假基因或推测的基因序列时("hypothetical protein" or "similar to..."),你可以选择PSI-BLAST重新试试。
3,PHI-BLAST : 模式发现迭代BLASTPHI-BLAST can do a restricted protein pattern search.PHI-BLAST, 模式发现迭代BLAST, 用蛋白查询来搜索蛋白数据库的一个程序。
仅仅找出那些查询序列中含有的特殊模式的对齐。
PHI的语法详细介绍看这里:/blast/html/PHIsyntax.htmlThe syntax for patterns in PHI-BLAST follows the conventions of PROSITE. When using the stand-alone program, it is permissible to have multiple patterns in a file separated by a blank line between patterns. When using the Web-page only one pattern is allowed per query.Valid protein characters for PHI-BLAST patterns:ABCDEFGHIKLMNPQRSTVWXYZUValid DNA characters for PHI-BLAST patterns:ACGTOther useful delimiters:[ ] means any one of the characters enclosed in the brackets e.g., [LFYT] means one occurrence of L or F or Y or T- means nothing (this is a spacer character used by PROSITE)x(5) means 5 positions in which any residue is allowed (and similarly for any other single number in parentheses after x)x(2,4) means 2 to 4 positions where any residue is allowed, and similarly for any other two numbers separated by a comma; the first number should be < the second number. can occur only at the end of a pattern and means nothing it may occur before a period (another spacer used by PROSITE) may be used at the end of the pattern and means nothing.When using the stand-alone program, the pattern should be in a file, with the first line starting:ID followed by 2 spaces and a text string giving the pattern a name. There should also be a line starting PA followed by 2 spaces followed by the pattern description.All other PROSITE codes in the first two columns are allowed, but only the HI code, described below is relevant to PHI-BLAST.Here is an example from PROSITE.ID CNMP_BINDING_2; PATTERN.AC PS00889;DT OCT-1993 (CREATED); OCT-1993 (DA TA UPDA TE); NOV-1995 (INFO UPDATE).DE Cyclic nucleotide-binding domain signature 2.PA [LIVMF]-G-E-x-[GAS]-[LIVM]-x(5,11)-R-[STAQ]-A-x-[LIVMA]-x-[STACV].NR /RELEASE=32,49340;NR /TOTAL=57(36); /POSITIVE=57(36); /UNKNOWN=0(0); /FALSE_POS=0(0);NR /FALSE_NEG=1; /PARTIAL=1;CC /TAXO-RANGE=??EP?; /MAX-REPEAT=2;The line starting ID gives the pattern a name. The lines starting AC, DT, DE, NR, NR, CC are relevant to PROSITE users, but irrelevant to PHI-BLAST. These lines are tolerated, but ignored by PHI-BLAST.The line starting PA describes the pattern as: one of LIVMF followed by G followed by E followed by any single character followed by one of GAS followed by one of LIVM followed by any 5 to 11 characters followed by R followed by one of STAQ followed by A followed by any single character followed by one of LIVMA followed by any single character followed by one of STACVIn this case the pattern ends with a period. It can end with nothing after the last specifying symbol or any number of > signs or periods or combination thereof.Here is another example, illustrating the use of an HI line.ID ER_TARGET; PATTERN. PA [KRHQSA]-[DENQ]-E-L>. HI (19 22) HI (201 204)In this example, the HI lines specify that the pattern occurs twice, once from positions 19 through 22 in the sequence and once from positions 201 through 204 in the sequence. These specifications are relevant when stand-alone HI-BLAST is used with the seedp option, in which the interesting occurrences of the pattern in the sequence are specified. In this case the HI lines specify which occurrence(s) of the pattern should be used to find good alignments.In general, the seedp option is more useful than the standard patternp option ONLY when the pattern occurs K > 1 times in the sequence AND the user is interested in matching to J < K of those occurrences. Then using the HI lines enables the user to specify which occurrences are of interest.Disclaimer Privacy statement1、基本概念相似性(Similarity)是指序列比对过程中用来描述检测序列和目标序列之间相同或相似碱基或氨基酸残基占全部比对碱基或氨基酸残基的比例的高低,属于量的判断。