Free Meson Spectral Functions on the Lattice
- 格式:pdf
- 大小:91.90 KB
- 文档页数:3
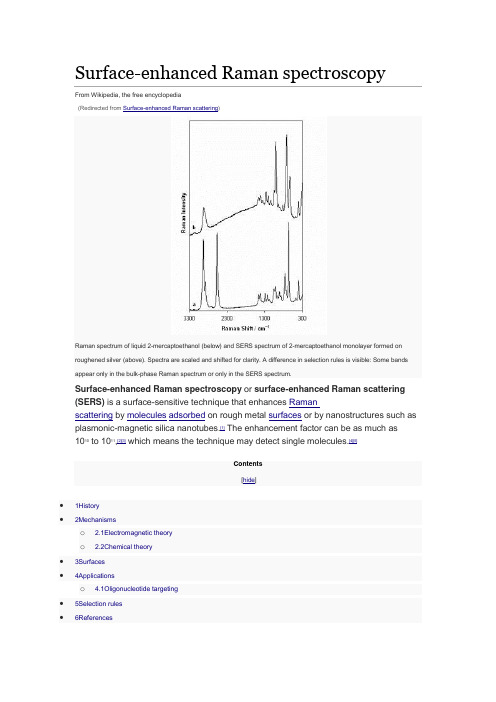
Surface-enhanced Raman spectroscopyFrom Wikipedia, the free encyclopedia(Redirected from Surface-enhanced Raman scattering)Raman spectrum of liquid 2-mercaptoethanol (below) and SERS spectrum of 2-mercaptoethanol monolayer formed on roughened silver (above). Spectra are scaled and shifted for clarity. A difference in selection rules is visible: Some bands appear only in the bulk-phase Raman spectrum or only in the SERS spectrum.Surface-enhanced Raman spectroscopy or surface-enhanced Raman scattering (SERS) is a surface-sensitive technique that enhances Ramanscattering by molecules adsorbed on rough metal surfaces or by nanostructures such as plasmonic-magnetic silica nanotubes.[1] The enhancement factor can be as much as 1010 to 1011,[2][3] which means the technique may detect single molecules.[4][5]Contents[hide]•1History•2Mechanismso 2.1Electromagnetic theoryo 2.2Chemical theory•3Surfaces•4Applicationso 4.1Oligonucleotide targeting•5Selection rules•6ReferencesHistory[edit]SERS from pyridine adsorbed on electrochemically roughened silver was first observed by Martin Fleischmann, Patrick J. Hendra and A. James McQuillan at the Department of Chemistry at the University of Southampton, Southampton, UK in 1973.[6] This initial publication has been cited over 4000 times. The 40th Anniversary of the first observation of the SERS effect has been marked by the Royal Society of Chemistry by the award of a National Chemical Landmark plaque to the University of Southampton. In 1977, two groups independently noted that the concentration of scattering species could not account for the enhanced signal and each proposed a mechanism for the observed enhancement. Their theories are still accepted as explaining the SERS effect. Jeanmaire and Van Duyne[7] proposed an electromagnetic effect, while Albrecht and Creighton[8] proposed a charge-transfer effect. Rufus Ritchie, of Oak Ridge National Laboratory's Health Sciences Research Division, predicted the existence of the surface plasmon.[9]Mechanisms[edit]The exact mechanism of the enhancement effect of SERS is still a matter of debate in the literature. There are two primary theories and while their mechanisms differ substantially, distinguishing them experimentally has not been straightforward.The electromagnetic theory proposes the excitation of localized surface plasmons, while the chemical theory proposes the formation of charge-transfer complexes. The chemical theory applies only for species that have formed a chemical bond with the surface, so it cannot explain the observed signal enhancement in all cases, whereas the electromagnetic theory can apply even in those cases where the specimenis physisorbed only to the surface. It has been shown recently that SERS enhancement can occur even when an excited molecule is relatively far apart from the surface which hosts metallic nanoparticles enabling surface plasmon phenomena.[10] This observation provides a strong support for the electromagnetic theory of SERS. Research in 2015 on a more powerful extension of the SERS technique called SLIPSERS (SlipperyLiquid-Infused Porous SERS)[11] has further supported the EM theory.[12] Electromagnetic theory[edit]The increase in intensity of the Raman signal for adsorbates on particular surfaces occurs because of an enhancement in the electric field provided by the surface. When the incident light in the experiment strikes the surface, localized surface plasmons are excited. The field enhancement is greatest when the plasmon frequency, ωp, is in resonance with the radiation. In order for scattering to occur, the plasmon oscillations must be perpendicular to the surface; if they are in-plane with the surface, no scattering will occur. It is because of this requirement that roughened surfaces or arrangementsof nanoparticles are typically employed in SERS experiments as these surfaces provide an area on which these localized collective oscillations can occur.[13]The light incident on the surface can excite a variety of phenomena in the surface, yet the complexity of this situation can be minimized by surfaces with features much smaller thanthe wavelength of the light, as only the dipolar contribution will be recognized by the system. The dipolar term contributes to the plasmon oscillations, which leads to the enhancement. The SERS effect is so pronounced because the field enhancement occurs twice. First, the field enhancement magnifies the intensity of incident light, which will excite the Raman modes of the molecule being studied, therefore increasing the signal of the Raman scattering. The Raman signal is then further magnified by the surface due to the same mechanism that excited the incident light, resulting in a greater increase in the total output. At each stage the electric field is enhanced as E2, for a total enhancement of E4.[14]The enhancement is not equal for all frequencies. For those frequencies for which the Raman signal is only slightly shifted from the incident light, both the incident laser light and the Raman signal can be near resonance with the plasmon frequency, leading to the E4 enhancement. When the frequency shift is large, the incident light and the Raman signal cannot both be on resonance with ωp, thus the enhancement at both stages cannot be maximal.[15]The choice of surface metal is also dictated by the plasmon resonancefrequency. Visible and near-infrared radiation (NIR) are used to excite Ramanmodes. Silver and gold are typical metals for SERS experiments because their plasmon resonance frequencies fall within these wavelength ranges, providing maximal enhancement for visible and NIR light. Copper's absorption spectrum also falls within the range acceptable for SERS experiments.[16] Platinum and palladium nanostructures also display plasmon resonance within visible and NIR frequencies.[17]Chemical theory[edit]While the electromagnetic theory of enhancement can be applied regardless of the molecule being studied, it does not fully explain the magnitude of the enhancement observed in many systems. For many molecules, often those with a lone pair of electrons, in which the molecules can bond to the surface, a different enhancement mechanism that does not involve surface plasmons has been described. This chemical mechanism involves charge transfer between the chemisorbed species and the metal surface. The chemical mechanism only applies in specific cases and probably occurs in concert with the electromagnetic mechanism.[18][19]The HOMO to LUMO transition for many molecules requires much more energy than the infrared or visible light typically involved in Raman experiments. When the HOMO and LUMO of the adsorbate fall symmetrically about the Fermi level of the metal surface, light of half the energy can be employed to make the transition, where the metal acts as a charge-transfer intermediate.[18] Thus a spectroscopic transition that might normally take place in the UV can be excited by visible light.[15]Surfaces[edit]While SERS can be performed in colloidal solutions, today the most common method for performing SERS measurements is by depositing a liquid sample onto a silicon or glasssurface with a nanostructured noble metal surface. While the first experiments were performed on electrochemically roughened silver,[20] now surfaces are often prepared using a distribution of metal nanoparticles on the surface[21] as well as usinglithography[22] or porous silicon as a support.[23][24]Applying a thin film of silver onto wafers of silicon, through a day-long immersion in a saturated solution of silver nitrate in n-octanol, is a widely accepted method to prepare a surface-enhanced Raman scattering substrate.[25] The most common metals used for plasmonic surfaces are silver and gold, however aluminum has recently been explored as an alternative plasmonic material, because its plasmon band is in the UV region, contrary to silver and gold.[26] Hence, there is great interest in using aluminum for UV SERS. It has, however, surprisingly also been shown to have a large enhancement in the infrared, which is not fully understood.[27] In the current decade, it has been recognized that the cost of SERS substrates must be reduced in order to become a commonly used analytical chemistry measurement technique.[28] To meet this need, plasmonic paper has experienced widespread attention in the field, with highly sensitive SERS substrates being formed through approaches such assoaking,[29][30][31] in-situ synthesis,[32][33] screen printing[34] and inkjet printing.[35][36][37]The shape and size of the metal nanoparticles strongly affect the strength of the enhancement because these factors influence the ratio of absorption and scattering events.[38][39] There is an ideal size for these particles, and an ideal surface thickness for each experiment.[40] Particles that are too large allow the excitation of multipoles, which are nonradiative. As only the dipole transition leads to Raman scattering, the higher-order transitions will cause a decrease in the overall efficiency of the enhancement. Particles that are too small lose their electrical conductance and cannot enhance the field. When the particle size approaches a few atoms, the definition of a plasmon does not hold, as there must be a large collection of electrons to oscillate together.[14] An ideal SERS substrate must possess high uniformity and high field enhancement. Such substrates can be fabricated on a wafer scale and label-free superresolution microscopy has also been demonstrated using the fluctuations of surface enhanced Raman scattering signal on such highly uniform, high-performance plasmonic metasurfaces. [41]Applications[edit]SERS substrates prepared with silver nano rods are used to detect the presence of low abundance biomolecules, and can therefore detect proteins in body fluids.[42][43][44] This technology has been utilized to detect urea and blood plasma label free in human serum and may become the next generation in cancer detection and screening.[43][44] The ability to analyze the composition of a mixture on the nano scale makes the use of SERS substrates beneficial for environmental analysis, pharmaceuticals, material sciences, art and archeological research, forensic science, drug and explosives detection, food quality analysis, and single algal cell detection.[45][46][47] SERS combined with plasmonic sensing can be used for high-sensitivity and quantitative detection of biomolecular interaction.[48]Oligonucleotide targeting[edit]SERS can be used to target specific DNA and RNA sequences using a combination of gold and silver nanoparticles and Raman-active dyes, such as Cy3. Specific single nucleotide polymorphisms (SNP) can be identified using this technique. The gold nanoparticles facilitate the formation of a silver coating on the dye-labeled regions of DNA or RNA, allowing SERS to be performed. This has several potential applications: For example, Cao et al. report that gene sequences for HIV, Ebola, Hepatitis, and Bacillus Anthracis can be uniquely identified using this technique. Each spectrum was specific, which is advantageous over fluorescence detection; some fluorescent markers overlap and interfere with other gene markers. The advantage of this technique to identify gene sequences is that several Raman dyes are commercially available, which could lead to the development of non-overlapping probes for gene detection.[49]Selection rules[edit]The term surface enhanced Raman spectroscopy implies that it provides the same information that traditional Raman spectroscopy does, simply with a greatly enhanced signal. While the spectra of most SERS experiments are similar to the non-surface enhanced spectra, there are often differences in the number of modes present. Additional modes not found in the traditional Raman spectrum can be present in the SERS spectrum, while other modes can disappear. The modes observed in any spectroscopic experiment are dictated by the symmetry of the molecules and are usually summarized by selection rules. When molecules are adsorbed to a surface, the symmetry of the system can change, slightly modifying the symmetry of the molecule, which can lead to differences in mode selection.[50]One common way in which selection rules are modified arises from the fact that many molecules that have a center of symmetry lose that feature when adsorbed to a surface. The loss of a center of symmetry eliminates the requirements of the mutual exclusion rule, which dictates that modes can only be either Raman or Infrared active. Thus modes that would normally appear only in the infrared spectrum of the free molecule can appear in the SERS spectrum.[13]A molecule's symmetry can be changed in different ways depending on the orientation in which the molecule is attached to the surface. In some experiments, it is possible to determine the orientation of adsorption to the surface from the SERS spectrum, as different modes will be present depending on how the symmetry is modified.[51]References[edit]1. Jump up^ Xu, X., Li, H., Hasan, D., Ruoff, R. S., Wang, A. X. and Fan, D. L. (2013), Near-Field EnhancedPlasmonic-Magnetic Bifunctional Nanotubes for Single Cell Bioanalysis. Adv. Funct.Mater.. doi:10.1002/adfm.2012038222. Jump up^Blackie, Evan J.; Le Ru, Eric C.; Etchegoin, Pablo G. (2009). "Single-Molecule Surface-EnhancedRaman Spectroscopy of Nonresonant Molecules". J. Am. Chem. Soc.131 (40):14466–14472. doi:10.1021/ja905319w.PMID19807188.3. Jump up^Blackie, Evan J.; Le Ru, Eric C.; Meyer, Matthias; Etchegoin, Pablo G. (2007). "Surface EnhancedRaman Scattering Enhancement Factors: A Comprehensive Study". J. Phys. Chem. C. 111 (37):13794–13803.doi:10.1021/jp0687908.4. Jump up^Nie, S; Emory, SR (1997). "Probing Single Molecules and Single Nanoparticles by Surface-EnhancedRaman Scattering". Science. 275 (5303): 1102–6.doi:10.1126/science.275.5303.1102. PMID9027306.5. Jump up^Le Ru, Eric C.; Meyer, Matthias; Etchegoin, Pablo G. (2006). "Proof of Single-Molecule Sensitivity inSurface Enhanced Raman Scattering (SERS) by Means of a Two-Analyte Technique". J. Phys. Chem. B. 110 (4): 1944–1948.doi:10.1021/jp054732v. PMID16471765.6. Jump up^Fleischmann, M.; PJ Hendra & AJ McQuillan (15 May 1974). "Raman Spectra of Pyridine Adsorbed ata Silver Electrode". Chemical Physics Letters. 26 (2):163–166.Bibcode:1974CPL....26..163F. doi:10.1016/0009-2614(74)85388-1.7. Jump up^Jeanmaire, David L.; Richard P. van Duyne (1977). "Surface Raman Electrochemistry Part I.Heterocyclic, Aromatic and Aliphatic Amines Adsorbed on the Anodized Silver Electrode". Journal ofElectroanalytical Chemistry.84: 1–20. doi:10.1016/S0022-0728(77)80224-6.8. Jump up^Albrecht, M. Grant; J. Alan Creighton (1977). "Anomalously Intense Raman Spectra of Pyridine at aSilver Electrode". Journal of the American Chemical Society. 99(15): 5215–5217. doi:10.1021/ja00457a071.9. Jump up^"Technical Highlights. New Probe Detects Trace Pollutants in Groundwater". Oak Ridge NationalLaboratory Review. 26 (2).10. Jump up^Kukushkin, V. I.; Van’kov, A. B.; Kukushkin, I. V. (2013). "Long-range manifestation ofsurface-enhanced Raman scattering". JETP Letters. 98 (2):64–69.doi:10.1134/S0021364013150113. ISSN0021-3640.11. Jump up^/content/early/2015/12/29/151898011312. Jumpup^/single-molecule-detection-of-contaminants-explosives-or-diseases-now-possible 13. ^ Jump up to:a b Smith, E.; Dent, G., Modern Raman Spectroscopy: A Practical Approach. John Wiley and Sons:2005 ISBN 0-471-49794-014. ^ Jump up to:a b Moskovits, M., Surface-Enhanced Raman Spectroscopy: a Brief Perspective. InSurface-Enhanced Raman Scattering – Physics and Applications, 2006; pp. 1–18ISBN 3-540-33566-815. ^ Jump up to:a b Campion, Alan; Kambhampati, Patanjali (1998). "Surface-enhanced Raman scattering". ChemicalSociety Reviews. 27 (4): 241. doi:10.1039/A827241Z.16. Jump up^Creighton, J. Alan; Eadon, Desmond G. (1991). "Ultraviolet?visible absorption spectra of the colloidalmetallic elements". Journal of the Chemical Society, Faraday Transactions. 87 (24):3881.doi:10.1039/FT9918703881.17. Jump up^Langhammer, Christoph; Yuan, Zhe; Zorić, Igor; Kasemo, Bengt (2006). "Plasmonic Properties ofSupported Pt and Pd Nanostructures". Nano Letters. 6 (4):833–838.Bibcode:2006NanoL...6..833L. doi:10.1021/nl060219x.PMID16608293.18. ^ Jump up to:a b Lombardi, John R.; Birke, Ronald L.; Lu, Tianhong; Xu, Jia (1986). "Charge-transfer theory ofsurface enhanced Raman spectroscopy: Herzberg–Teller contributions". The Journal of Chemical Physics. 84 (8): 4174. Bibcode:1986JChPh..84.4174L.doi:10.1063/1.450037.19. Jump up^Lombardi, J.R.; Birke, R.L. (2008). "A Unified Approach to Surface-Enhanced RamanSpectroscopy". Journal of Physical Chemistry C. 112 (14): 5605–5617.doi:10.1021/jp800167v.20. Jump up^Fleischmann, M.; Hendra, P.J.; McQuillan, A.J. (1974). "Raman spectra of pyridine adsorbed at a silverelectrode". Chemical Physics Letters. 26 (2):163–166.Bibcode:1974CPL....26..163F. doi:10.1016/0009-2614(74)85388-1.21. Jump up^Mock, J. J.; Barbic, M.; Smith, D. R.; Schultz, D. A.; Schultz, S. (2002). "Shape effects in plasmonresonance of individual colloidal silver nanoparticles". The Journal of Chemical Physics. 116 (15):6755.Bibcode:2002JChPh.116.6755M. doi:10.1063/1.1462610.22. Jump up^Witlicki, Edward H.; et al. (2011). "Molecular Logic Gates Using Surface-Enhanced Raman-ScatteredLight". J. Am. Chem. Soc.133 (19): 7288–7291.doi:10.1021/ja200992x.23. Jump up^Lin, Haohao; Mock, Jack; Smith, David; Gao, Ting; Sailor, Michael J. (August 2004)."Surface-Enhanced Raman Scattering from Silver-Plated Porous Silicon". The Journal of Physical ChemistryB. 108 (31): 11654–11659.doi:10.1021/jp049008b.24. Jump up^Talian, Ivan; Mogensen, Klaus Bo; Oriňák, Andrej; Kaniansky, Dušan; Hübner, Jörg (August 2009)."Surface-enhanced Raman spectroscopy on novel black silicon-based nanostructured surfaces". Journal ofRaman Spectroscopy.40 (8): 982–986. doi:10.1002/jrs.2213.25. Jump up^Shrestha, LK; Wi JS; Williams J; Akada M; Ariga K (March 2014). "Facile fabrication of silvernanoclusters as promising surface-enhanced Raman scattering substrates".Journal of Nanoscience andNanotechnology. 14 (3): 2245–51. doi:10.1166/jnn.2014.8538. PMID24745219.26. Jump up^Dörfer, Thomas; Schmitt, Michael; Popp, Jürgen (November 2007). "Deep-UV surface-enhancedRaman scattering".Journal of Raman Spectroscopy. 38 (11): 1379–1382.doi:10.1002/jrs.1831.27. Jump up^Mogensen, Klaus Bo; Gühlke, Marina; Kneipp, Janina; Kadkhodazadeh, Shima; Wagner, Jakob B.;Espina Palanco, Marta; Kneipp, Harald; Kneipp, Katrin (2014). "Surface-enhanced Raman scattering on aluminum using near infrared and visible excitation". Chemical Communications. 50 (28): 3744. doi:10.1039/c4cc00010b. 28. Jump up^Hoppmann, Eric P.; Yu, Wei W.; White, Ian M. (2014)."Inkjet-Printed Fluidic Paper Devices forChemical and Biological Analytics Using Surface Enhanced Raman spectroscopy"(PDF). IEEE. IEEE. 20 (3): 195–204.doi:10.1109/jstqe.2013.2286076.29. Jump up^Lee, Chang H.; Tian, Limei; Singamaneni, Srikanth (2010). "Paper-Based SERS". ACS. AmericanChemical Society. 2 (12): 3429–3435. doi:10.1021/am1009875. Retrieved 2015-01-16.30. Jump up^Ngo, Ying Hui; Li, Dan; Simon, George P.; Garnier, Gil (2012). "Gold Nanoparticle". Langmuir.American Chemical Society. 28 (23): 8782–8790. doi:10.1021/la3012734. Retrieved 2015-01-16.31. Jump up^Ngo, Ying Hui; Li, Dan; Simon, George P.; Garnier, Gil (2013). "Effect of cationic polyacrylamides onthe aggregation and SERS". Journal of Colloid and Interface Science. Elsevier. 392:237–246.doi:10.1016/j.jcis.2012.09.080.32. Jump up^Laserna, J. J.; Campiglia, A. D.; Winefordner, J. D. (1989). "Mixture analysis and quantitativedetermination of nitrogen-containing organic molecules by surface-enhanced Raman spectrometry". Anal. Chem.American Chemical Society. 61 (15): 1697–1701.doi:10.1021/ac00190a022.33. Jump up^Chang, Yung; Yandi, Wetra; Chen, Wen-Yih; Shih, Yu-Ju; Yang, Chang-Chung; Chang, Yu; Ling,Qing-Dong; Higuchi, Akon (2010). "Tunable Bioadhesive Copolymer Hydrogels of Thermoresponsive Poly( N -isopropyl acrylamide) Containing Zwitterionic Polysulfobetaine". Biomacromolecules. American ChemicalSociety. 11 (4): 1101–1110.doi:10.1021/bm100093g.34. Jump up^Qu, Lu-Lu; Li, Da-Wei; Xue, Jin-Qun; Zhai, Wen-Lei; Fossey, John S.; Long, Yi-Tao(2012-02-07). "Batch fabrication of disposable screen printed SERS arrays".Lab Chip. 12 (5):876–881. doi:10.1039/C2LC20926H.ISSN1473-0189.35. Jump up^Yu, Wei W.; White, Ian M. (2013). "Inkjet-printed paper-based SERS". Analyst. Royal Society ofChemistry. 138(4): 1020. doi:10.1039/c2an36116g.36. Jump up^Hoppmann, Eric P.; Yu, Wei W.; White, Ian M. (2013)."Highly sensitive and flexible inkjet printedSERS"(PDF). Methods. Elsevier. 63 (3): 219–224.doi:10.1016/j.ymeth.2013.07.010.37. Jump up^Fierro-Mercado, Pedro M.; Hern, Samuel P. (2012)."Highly Sensitive Filter Paper Substrate forSERS".International Journal of Spectroscopy. Hindawi Publishing Corporation. 2012:1–7. doi:10.1155/2012/716527.38. Jump up^H. Lu; Zhang, Haixi; Yu, Xia; Zeng, Shuwen; Yong, Ken-Tye; Ho, Ho-Pui (2011). "Seed-mediatedPlasmon-driven Regrowth of Silver Nanodecahedrons (NDs)"(PDF).Plasmonics. 7 (1):167–173. doi:10.1007/s11468-011-9290-8.39. Jump up^ Aroca, R., Surface-enhanced Vibrational Spectroscopy. John Wiley & Sons (2006) ISBN0-471-60731-240. Jump up^Bao, Li-Li; Mahurin, Shannon M.; Liang, Cheng-Du; Dai, Sheng (2003). "Study of silver films over silicabeads as a surface-enhanced Raman scattering (SERS) substrate for detection of benzoic acid". Journal of Raman Spectroscopy.34 (5): 394–398. Bibcode:2003JRSp...34..394B.doi:10.1002/jrs.993.41. Jump up^Ayas, S. (2013). "Label-Free Nanometer-Resolution Imaging of Biological Architectures throughSurface Enhanced Raman Scattering". Scientific Reports. 3: 2624.doi:10.1038/srep02624.doi:10.1038/srep02624 42. Jump up^Yang, J; et al. (May 2013). "Surface-Enhanced Raman Spectroscopy Based Quantitative Bioassay onAptamer-Functionalized Nanopillars Using Large-Area Raman Mapping"(PDF). ACS Nano. 7 (6):5350–5359.doi:10.1021/nn401199k.43. ^ Jump up to:a b Han, YA; Ju J; Yoon Y; Kim SM (May 2014)."Fabrication of cost-effective surface enhancedRaman spectroscopy substrate using glancing angle deposition for the detection of urea in body fluid". Journal of Nanoscience and Nanotechnology. 14 (5): 3797–9.doi:10.1166/jnn.2014.8184. PMID24734638.44. ^ Jump up to:a b Li, D; Feng S; Huang H; Chen W; Shi H; Liu N; Chen L; Chen W; Yu Y; Chen R (March2014). "Label-free detection of blood plasma using silver nanoparticle based surface-enhanced Ramanspectroscopy for esophageal cancer screening". Journal of Nanoscience and Nanotechnology.10 (3):478–84. doi:10.1166/jbn.2014.1750.PMID24730243.45. Jump up^Deng, Y; Juang Y (March 2014). "Black silicon SERS substrate: Effect of surface morphology on SERSdetection and application of single algal cell analysis".Biosensors and Bioelectronics. 53:37–42.doi:10.1016/j.bios.2013.09.032.46. Jump up^Hoppmann, Eric; et al. (2013). Trace detection overcoming the cost and usability limitations oftraditional SERS technology(PDF) (Technical report). Diagnostic anSERS.line feed characterin |title= at position 50 (help)47. Jump up^Wackerbarth H; Salb C; Gundrum L; Niederkrüger M; Christou K; Beushausen V; Viöl W(2010). "Detection of explosives based on surface-enhanced Raman spectroscopy". Applied Optics. 49 (23): 4362–4366.doi:10.1364/AO.49.004362.48. Jump up^Xu, Zhida; Jiang, Jing; Wang, Xinhao; Han, Kevin; Ameen, Abid; Khan, Ibrahim; Chang, Te-Wei; Liu,Logan (2016)."Large-area, uniform and low-cost dual-mode plasmonic naked-eye colorimetry and SERS sensor with handheld Raman spectrometer". Nanoscale. 8: 6162–6172.doi:10.1039/C5NR08357E.49. Jump up^Cao, Y. C.; Jin, R; Mirkin, CA (2002). "Nanoparticles with Raman Spectroscopic Fingerprints for DNAand RNA Detection". Science. 297 (5586):1536–1540.Bibcode:2002Sci...297.1536C.doi:10.1126/science.297.5586.1536. PMID12202825.50. Jump up^Moskovits, M.; Suh, J. S. (1984). "Surface selection rules for surface-enhanced Raman spectroscopy:calculations and application to the surface-enhanced Raman spectrum of phthalazine on silver". The Journal of Physical Chemistry. 88 (23): 5526–5530.doi:10.1021/j150667a013.51. Jump up^Brolo, A.G.; Jiang, Z.; Irish, D.E. (2003). "The orientation of 2,2′-bipyridine adsorbed at a SERS-activeAu(111) electrode surface"(PDF). Journal of Electroanalytical Chemistry. 547 (2):163–172.doi:10.1016/S0022-0728(03)00215-8.。



第39卷第1期2021年2月辐射研究与辐射工艺学报J.Radiat.Res.Radiat.Process./fushe/CN/volumn/home.shtmlV ol.39 No.1February2021溶剂化电子研究的前沿进展胡长江马骏(南京航空航天大学材料科学与技术学院南京230026)摘要溶剂化电子是自然界中最小的阴离子和最强的还原性粒子,也是辐射化学反应过程中重要的活性物质。
溶剂化电子的研究将为溶液自由基反应、乏燃料后处理中溶剂与萃取剂的辐射化学、生命过程的电荷转移与电荷传输等领域提供关键信息,因此是有机化学、无机化学、辐射化学和放射生物学研究中的重要课题。
近年来,随着短脉冲激光技术的新一轮革命,有关溶剂化电子的研究迎来了新的一轮爆发增长期。
为此,本文力图概述当前溶剂化电子研究的前沿进展,主要内容涉及溶剂化电子结合能的测定、溶剂化电子的表面态以及预溶剂化电子和准自由电子与核苷酸分子的反应动力学等。
关键词溶剂化电子,结合能,液相光电子能谱法,脉冲辐解中图分类号TL13DOI:10.11889/j.1000-3436.2021.rrj.39.010101An overview of solvated electrons:recent advancesHU Changjiang MA Jun(College of Materials Science and Technology,Nanjing University of Aeronautics and Astronautics,Nanjing230026,China)ABSTRACT Solvated electrons are the smallest and most reductive particles in nature,and also are the importantreactive species in radiation chemistry.The study of solvated electrons will provide key information for the field offree-radical reactions,the radiation effects of solvents and extractants in spent fuel post-treatment,and electrontransfer and transport occuring in life activities etc.Therefore,it has been a subject of intense interests in disciplinesof organic chemistry,inorganic chemistry,radiation chemistry and radiation biology.Owning to the accelaratedadvances achievied in ultrashort pulse laser technology,there has been an increasing understanding solvated electronin recent years.This paper will briefly introduce the context of these updated knowledges.It includes the study ofdetermination of the binding energy of solvated electrons,the surface-bound states of solvated electrons,and thekinetics of pre-solvated electrons and quasi-free electrons with nucleotide molecules in solutions.KEYWORDS Solvated electron,Binding energy,Photoelectron spectroscopy,Pulse radiolysisCLC TL13溶剂化电子(Solvated electron,esol‒)是与其周围溶剂分子形成平衡态构型的定域化(陷落)基金资助:国家自然科学基金(11975122、21906083)和江苏省自然科学基金(BK2019030384)资助第一作者:胡长江,男,1993年7月出生,2019年于三峡大学材料与化工学院获硕士学位,现为南京航空航天大学博士研究生通信作者:马骏,博士,教授,E-mail:**************.cn收稿日期:初稿2020-09-25;修回2020-11-20Supported by National Natural Science Foundation of China(11975122,21906083)and Natural Science Foundation of Jiangsu Province(BK2019030384)First author:HU Changjiang(male)was born in July1993,and obtainted his master’s degree from College of Materials and Chemical Engineering,Three Gorges University in2019.Now he is a graduate student at Nanjing University of Aeronautics and AstronauticsCorresponding author:MA Jun,doctoral degree,professor,E-mail:**************.cnReceived25September2020;accepted20November2020辐射研究与辐射工艺学报2021 39:010101电子。

M O L C A S:分子激发态Zork译自:《MOLCAS version 5.2: MOLCAS MANUAL》准确计算分子激发态才成为可能。
由于CASPT2我们选择噻吩(thiophene)1.2 产生里德堡基函数1.3 SEWARD和CASSCF计算1.4 CASPT2计算1.5 跃迁偶极矩计算二. 里德堡轨道和里德堡态的影响。
一个例子:鸟嘌呤三. 其它的例子 参考文献一. 噻吩的垂直光谱由于系统大小以及计算的积分,不仅通常的典型从头计算程序存在限制,CASPT2方法也有尺寸以及在初始CASSCF 步骤中选择活性空间的基本限制。
不仅是因为空间不能太大,还因为活性空间定义了多重组态波函中的组态类型和数量。
描述所有态的近简并组态必须出现在参考波函中。
因此,清楚了解研究的体系,对于计算的设计是非常必要的。
对激发态来说,这将限制可以研究的电子态的数量。
1.1 计算的设计 1.2 产生里德堡基函数 1.3 SEWARD 和CASSCF 计算1.4 CASPT2计算 1.5 跃迁偶极矩计算噻吩是一个平面五体环分子,包括一个硫原子和四个碳原子。
体系的π结构包括碳原子之间的两个共轭双键。
因此,轨道的π价电子结构由两个π键,两个π*反键轨道,以及位于硫原子上的一个非键π轨道构成。
在此类体系中,π轨道是最高占据轨道,由该轨道的激发形成了气体和溶液的紫外-可见光谱。
同样,包含在低位激发态中典型的轨道也有孤对轨道,例如与分子的σ轮廓图位于同一平面的硫原子n 轨道。
另一方面,形成C-C 和C-H 键的σ轨道不参与形成低位电子激发态。
图1. 噻吩考虑到这些,我们必须把三个最低的π和两个π*价轨道,以及硫原子上的σ孤对价轨道包含到活性空间中。
分子属于C2v点群,因此有三个b1,两个a2的π,π*轨道,以及一个a1的n轨道。
也就是说,我们最小的价活性空间标记为(1302),其中的每一个数字分别对应a1, a2, b1和b2轨道。

a rXiv:h ep-ph/949288v113Se p1994Preprint IHEP 94-92September 1994Submitted to JETP Lett.αS from Spectroscopy of ψ-and Υ-particles in QCD Sum Rules V.V.Kiselev Institute for High Energy Physics,Protvino,Moscow Region,142284,Russia,E-mail:kiselev@mx.ihep.su Fax:+7-095-230-23-37Abstract In a specific scheme of the QCD sum rules,one gives the estimate of αS ≃0.20from the data on the masses and leptonic constants of ψ-and Υ-particles.The characteristic value of the quark-gluon coupling in QCD for the systems of the charmonium (¯c c )and bottomonium (¯bb )is generally estimated from the branch-ing fractions of the radiative and leptonic decays of vector states Br (Q ¯Q →γX )≃Γ(Q ¯Q →γgg )/Γ(Q ¯Q →ggg ),Br (Q ¯Q →l +l −)≃Γ(Q ¯Q →l +l −)/Γ(Q ¯Q →ggg )[1],so these ways do not depend on a modelling of the heavy quarkonium wave function.However,such estimates contain the uncertainty,related with a model for the gluon hadronization.For instance,one can think,that the gluons have a nonzero virtuality of the order of the confinement scale or higher corrections generatea nonzero effective dynamical mass of the gluon [2].The αS estimates from the total cross sections for the hadronic production of the c -andb -quarks also contain the un-certainties,related with an account of higher corrections in the perturbation theory of QCD (the K -factor),a choice of the quark mass values and a model-dependence of the parton distributions.Therefore,one generally supposes αS (ψ,Υ)≃0.3÷0.2.It would be useful one to obtain the characteristic αS value for the ψ-and Υ-particles,using another way of the estimate.To get the αS estimate,in the present paper we use the relations,obtained in the framework of the QCD sum rules [3]for the leptonic constants and masses of the heavy quarkonia.In the recently offered scheme of the QCD sum rules [4],in the leading approxi-mation over the inverse heavy quark mass and with an account of the coulomb-like αS /v -corrections,one has got the relations for the leptonic constants f n of the heavy quarkonium nS -levels,lying below the threshold of the decay into the heavy meson pair,[4]f 2n πdM nand for the difference of the level masses[5]dM nM n−M1==const.,(3)MandM n−M1ln2M(1S)5.V.V.Kiselev,Preprint IHEP94-74,Protvino(1994).6.E.Eichten,Preprint FERMILAB-Conf-85/29-T(1985);C.Quigg and J.L.Rosner,Phys.Lett.B71,153(1977).3。
基于HITEMP数据库的分子吸收光谱高精度快速建模方法钱宝健,蔡静*,常海涛,高一凡(航空工业北京长城计量测试技术研究所,北京 100095)摘要:为解决高温环境下分子吸收光谱精确计算的时间复杂性,满足宽光谱测量领域对理论吸收光谱计算的需求,本研究利用Python语言以逐线计算为基础,结合线型函数的简化、线翼截止准则和谱线数据库的优化,建立了基于高温分子吸收参数数据库(High⁃Temperature molecular spectroscopic absorption parameters data⁃base,HITEMP)的分子吸收光谱精确快速计算模型。
以Hartmann⁃Tran线型函数作为吸收光谱标准线型编写部分相关二次速度依赖硬碰撞函数(partially⁃Correlated quadratic⁃Speed⁃Dependent Hard⁃Collision Profile,pCqSDHC),结合复概率函数(Complex Probability Function,CPF)简化模型实现了线型函数的精确快速计算,相较于理论计算模型计算速度提高了20倍。
按照光谱计算残差在10-5量级确定了固定波数截断结合谱线半宽等倍数截断的线翼截止准则。
以阈值线强度10-25 cm-1/(mol∙cm-2)为标准筛选了每100 K温度梯度时的光谱数据,整合得到优化数据库。
在6 500 ~ 8 000 cm-1范围内对水分子的吸收光谱进行计算,并与“”分子气体集成光谱建模网站仿真结果对比,逐线模型的计算误差在10-7量级,优化模型的计算误差在10-5量级,计算速度平均提升25倍。
该模型满足吸收光谱测量中对于理论吸收光谱的高效准确计算,为复杂环境中基于宽调谐、超连续激光吸收光谱的测量研究提供了理论模型基础。
关键词:吸收光谱;HITEMP数据库;线型函数;线翼截止中图分类号:TB9;O433 文献标志码:A 文章编号:1674-5795(2023)05-0039-10Modeling molecular absorption spectra based on the HITEMP databaseQIAN Baojian, CAI Jing*, CHANG Haitao, GAO Yifan(Changcheng Institute of Metrology & Measurement, Beijing 100095, China)Abstract: To address the computational complexity of accurately calculating molecular absorption spectra in high⁃temperature environments and meet the demand for theoretical absorption spectrum calculations in broad⁃spectrum mea⁃surement fields, this study developed a precise and fast calculation model for molecular absorption spectra based on the High⁃Temperature molecular spectroscopic absorption parameters database (HITEMP). The model was implemented us⁃ing Python language, employing a line⁃by⁃line calculation approach combined with simplification of line shape functions, line wing truncation criteria, and optimization of spectral line databases. The Hartmann⁃Tran line shape function was used as the standard absorption spectrum line shape, and partially⁃Correlated quadratic⁃Speed⁃Dependent Hard⁃Collision Pro⁃file (pCqSDHC) was developed for relevant second⁃order velocity⁃dependent hard⁃collision functions. By incorporating the doi:10.11823/j.issn.1674-5795.2023.05.06收稿日期:2023-09-26;修回日期:2023-10-08基金项目:国家“十三五”计量技术基础科研项目(JSJL2020205A003)引用格式:钱宝健,蔡静,常海涛,等.基于HITEMP数据库的分子吸收光谱高精度快速建模方法[J].计测技术,2023,43(5):39-48.Citation:QIAN B J,CAI J,CHANG H T,et al.Modeling molecular absorption spectra based on the HITEMP database[J].Metrology & Measurement Technology,2023,43(5):39-48.Complex Probability Function (CPF) and simplifying the model, the line shape functions were calculated accurately and rapidly, resulting in a 20⁃fold increase in computational speed compared to theoretical models. The line wing truncation criteria were determined based on the spectral calculation residual at the level of 10-5 and involved the truncation of fixed wavenumbers combined with equal multiple truncations of spectral line half widths. Spectral data for each temperature gradient of 100 K were selected using a threshold line intensity of 10-25 cm-1/(mol∙cm-2) and integrated to create an opti⁃mized database. The absorption spectra of water molecules were calculated within the range of 6 500 ~ 8 000 cm-1 and compared with the simulation results from "", a molecular gas integrated spectral modeling website. The calculation error of the line⁃by⁃line model was at the level of 10-7, while the optimized model achieved a calculation error at the level of 10-5, with an average speed improvement of 25 times. This model enables efficient and accurate calculation of theoretical absorption spectra for absorption spectral measurements and provides a theoretical foundation for measuring studies based on wide⁃tunable and supercontinuum laser absorption spectra in complex environments.Key words: absorption spectrum; HITEMP database; line shape functions; line wing cutoff0 引言分子吸收光谱是一种描述物质分子对特定波长光的吸收能力的图谱,通过测量物质对不同波长光的吸收程度,可以推断物质的组成、浓度、结构和化学性质等重要信息,从而在燃烧诊断[1-2]、温度测量[3-4]、污染物监测[5]等领域中进行定性和定量分析。
纳米金放大SPR技术检测前列腺癌肿瘤标志物free PSA 陈胜华;王文武;向娟【摘要】表面等离子体激元共振(surface plasmon resonance,SPR)是一种基于芯片表面折射率变化的光学检测技术,具有快速、灵敏、免标记等优点,可实时在线地跟踪固液界面反应,在蛋白及核酸等生物分子检测方面具有广阔的应用前景。
但是,传统的SPR检测方法难以检测低分子量、超低浓度的生物分子。
纳米金具有密度大、介电常数大、良好的生物相容性等特点,因此,基于纳米金放大的SPR技术既可以保持生物分子的生物活性,又可以有效突破传统SPR中检测限的限制。
游离前列腺特异性抗原(free prostate-specific antigenf,-PSA)是一种常用于前列腺癌早期诊断的标志物,但由于其在血清中的浓度非常低,给前列腺癌的早期诊断造成困难。
本论文利用基于纳米金放大的SPR技术,设计竞争型免疫模式实现了f-PSA 的超灵敏检测。
%Surface plasmon resonance(SPR) is an optical analytical technique based on the ability to detect dielectric constant changes induced by molecular adsorption on a noble metal film.SPR is attractive owing to several inherent advantages,such as label-free analysis,rapid and high sensitivity and monitor interfacial reactions at solid-liquid interface.Unfortunately,the inability of conventional SPR to measure extremely small changes in refractive index hinders its application in ultra sensitive detection.Gold nanoparticles(AuNPs) have been used to strengthen the SPR signal due to the ability of their high mass,large dielectric constant,and good biocompatibility.Free prostate-specific antigen(f-PSA) is an effective serum marker currently available for the detection of prostate cancer.Detection of low concentration f-PSA isimportant to improve the accuracy of the early diagnosis of prostate cancer.In this paper,competitive immunoassay was designed for ultra sensitive determination of f-PSA based on the technology of AuNPs amplified the SPR response.【期刊名称】《中州大学学报》【年(卷),期】2011(028)005【总页数】4页(P115-118)【关键词】表面等离子体激元共振;游离前列腺特异性抗原;纳米金放大;免疫反应【作者】陈胜华;王文武;向娟【作者单位】中南大学化学化工学院,长沙410083;中州大学化工食品学院,郑州450044;中南大学化学化工学院,长沙410083【正文语种】中文【中图分类】TG1461.绪论肿瘤标志物检测是肿瘤普查、诊断和预后的主要技术手段之一。
a r X i v :h e p -p h /0510277v 2 13 J a n 2006D +sJ (2317)→D +s π0decay widthMarina NielsenInstituto de F´ısica,Universidade de S˜a o Paulo,C.P.66318,05389-970S˜a o Paulo,SP,BrazilWe use the QCD sum rules to analize the hadronic decay D +sJ (2317)→D +s π0,in the hypotesisthat the D +sJ (2317)can be identified as a four-quark state.We use a diquak-antidiquark current and work to the order of m s in full QCD,without relying on 1/m c expansion.We find that the partial decay width of the hadronic isospin violating mode is proportional to the isovector quarkcondensate, 0|¯dd −¯u u |0 .The estimated partial decay width is of the order of 6keV.PACS numbers:11.55.Hx,12.38.Lg ,13.25.-kThe strange-charmed mesons D +sJ (2317)and D +sJ (2460)with spin parity 0+,1+[1,2,3,4]are lighter than the prediction of the very successful quark model for the charmed mesons [5].One interpretation is that this is evidence for a chiral symmetry where the mass gap between the 0−and 1−states equates the mass gap between the 0+and 1+states [6].Other interpretations are related with the underlying structure of these mesons,which has been extensively debated.They have been interpreted as conventional c ¯s states [7,8,9,10,11,12,13,14,15,16],two-meson molecular state [17,18],D −K -mixing [19]or four-quark states [20,21,22,23,24,25,26,27].Because of their low masses,these two states are lower than the DK and D ∗K thresholds.Therefore,their strong decays must proceed through isospin violating effects.There have been some discussions of their decays within the quark model [6,13,14,15,20]and QCD sum rules [16].In all these studies but [14],the isospin violating effects were considered through the η−π0mixing.However,if these mesons are considered as four-quark states,in a QCD sum rule calculation,the isospin violating effects can be introduced through the mass and quark condensate difference between the u and d quarks.In a recent calculation [23]the scalar-isoscalar meson D +sJ (2317)were considered as a S -wave bound state of a diquark-antidiquark pair.As suggested in ref.[28],the diquark was taken to be a spin zero colour anti-triplet.The corresponding interpolating field is:j S =ǫabc ǫdec 2(u T a Cγ5c b )(¯u d γ5C ¯s T e )+(d T a Cγ5c b )(¯d d γ5C ¯s T e) ,(1)where a,b,c,...are colour indices and C is the charge conjugation matrix.In ref.[23],using the QCD sum rule (QCDSR)formalism [29,30,31],it was shown that it is possible to reproduce the experimental mass of the meson D +sJ (2317)using this four-quark state picture.Here,we extend the calculation done in ref.[23]to study the vertexassociated with the decay D +sJ (2317)→D +s π0.The QCDSR calculation for the vertex,D +sJ (2317)D +s π0,centers around the three-point function given byΓµ(p,p ′,q )=d 4xd 4ye ip ′.x e iq.y 0|T [j D s (x )j π05µ(y )j †S (0)]|0 ,(2)where p =p ′+q and the interpolating fields for the pion and D s mesons are given by:j π5µ=12(¯u a γµγ5u a −¯da γµγ5d a ),j D s =i ¯s a γ5c a .(3)The fundamental assumption of the QCD sum rule approach is the principle of duality.Specifically,we assumethat there is an interval over which the above vertex function may be equivalently described at both,the quark level and at the hadron level.Therefore,the underlying procedure of the QCD sum rule technique is the following:on one hand we calculate the vertex function at the quark level in terms of quark and gluon fields.On the other hand,the vertex function is calculated at the hadronic level introducing hadron characteristics such as masses and coupling constants.At the quark level the complex structure of the QCD vacuum leads us to employ the Wilson’s operator product expansion (OPE).The calculation of the phenomenological side proceeds by inserting intermediate states for D s ,π0and D sJ ,and by using the definitions:0|j π05µ|π0(q )=iq µF π,0|j D s |D s (p ′) =m 2D s f D s(m c +m s )(p 2−m 2D sJ )(p ′2−m 2D s )(q 2−m 2π)q µ+continuum contribution ,(5)where the coupling constant g D sJ D s πis defined by the on-mass-shell matrix elementD s π|D sJ =g D sJ D s π.(6)The continuum contribution in Eq.(5)contains the contributions of all possible excited states.For the light scalar mesons,considered as diquark-antidiquark states,the study of their vertices functions using the QCD sum rule approach at the pion pole [11,30,32,33],was done in ref.[34].In Table I we show the results obtained for the different vertices studied in ref.[34],as well as the experimental values.Table I:Numerical results for the coupling constantsvertexg exp (GeV)3.1±0.5κK +π−4.5±0.41.6±0.1f 0π+π−1.6±0.8From Table I we see that,although not exactly in between the experimental error bars,the hadronic couplings determined from the QCD sum rule calculation are consistent with existing experimental data.The biggest discrepancy is for g f 0π+π−and this can be understood since the f 0→π+π−decay is mediated by gluon exchange and,therefore,probably in this case αs corrections,which were not considered,could play an important role.Here,we follow ref.[34]and work at the pion pole.The main reason for working at the pion pole is that the matrix element in Eq.(6)defines the coupling constant only at the pion pole.For q 2=0one would have to replace g D sJ D s π,in Eq.(6),by the form factor g D sJ D s π(q 2)and,therefore,one would have to deal with the complications associated with the extrapolation of the form factor [35,36].The pion pole method consists in neglecting the pion mass in the denominator of Eq.(5)and working at q 2=0.In the OPE side one singles out the leading terms in the operatorproduct expansion of Eq.(2)that match the 1/q 2term.In the phenomenological side,in the structure qµ(m c +m s )(p 2−m 2D sJ )(p ′2−m 2D s )+∞m 2cρcont (p 2,u )s 0−p 2Θ(u −u 0)[37,38],with s 0and u 0being the continuum thresholds for D sJ and D s respectively.Since we are working at q 2=0,we take the limit p 2=p ′2and we apply the Borel transformation to p 2→M 2to get:Γphen(M 2)=λS m 2D s f D s F πg D sJ D s π(s 0−u )du,and ρcc (u )=b (u)where the spectral density,ρOP E ,is given by the imaginary part of the correlation function.Transferring the pure continuum contribution to the OPE side,the sum rule for the coupling constant,up to dimension 7,is given by:C e −m 2D s /M 2−e −m 2D sJ/M2 +A e −s 0/M 2=γ ¯q q 1u2+m c m s u −m c ß12e−m 2c /M 21+m 2c (m c +m s )(m 2D sJ −m 2D s )g D sJ D s π.(12)InEq.(11),γmeasures theisospin symmetrybreaking in the quark condensate:γ=0|¯dd −¯u u |0ss =0.8 ¯q q .For the meson decay constants we use F π=√s 0≤2.8GeV we get λs =(2.9±0.3)×10−3GeV 5.In Fig.1we show,through the dots,the right-hand side (RHS)of Eq.(11),for u 0=6GeV 2and γ=−1×10−2[40],as a function of the Borel mass.We use the same Borel window as defined in ref.[23].1 1.21.4 1.61.82M 2(GeV 2)2e−074e−076e−078e−07R H S X L H S (G e V 7)FIG.1:Dots:the RHS of Eq.(11),as a function of the Borel mass.The solid line gives the fit of the QCDSR results through the LHS of Eq.(11).To determine g D sJ D s πwe fit the QCDSR results with the analytical expression in the left-hand side (LHS)of Eq.(11):C e −m 2D s /M 2−e −m 2D sJ /M 2 +A e −s 0/M 2,(14)and we get (using√s 0=2.7GeV)we get g D sJ D s π=50MeV.Allowings 0to vary in the interval 2.6≤√s 0=2.7GeV and varying the quark condensate,the charm quark and the strange quark masses in theintervals:−(0.24)3≤ ¯q q ≤−(0.22)3GeV 3,1.1≤m c ≤1.3GeV and 0.11≤m s ≤0.15GeV,we get results for the coupling constant still between the lower and upper limits given above.However,varying the value of γform −1×10−2to the more recent value given in [41]:γ=−2.6×10−2,and keeping the other parameters fixed we get g D sJ D s π=130MeV.On the other hand,if we use the smallest (in module)value allowed for γ:γ=−2.×10−3,we get g D sJ D s π=10MeV.Therefore,the biggest source of uncertainty in our calculation is the value of γ.In all cases considered here,the quality of the fit between the LHS and the RHS of Eq.(11)is similar to the one shown in Fig.1.The coupling constant,g D sJ D s π,is related with the partial decay width through the relation:Γ(D +sJ (2317)→D +s π0)=1λ(m 2D sJ ,m 2D s ,m 2π),(15)where λ(a,b,c )=a 2+b 2+c 2−2ab −2ac −2bc .Considering γ=−1×10−2,which was the value found in [40]tobe consistent with the neutron-proton mass difference in a QCDSR calculation,and allowing ¯q q ,m c ,m s and s 0to vary in the ranges discussed above we get:Γ(D +sJ (2317)→D +s π0)=(6±2)keV .(16)However,it is important to state that,if the value for γfound in ref.[41]proves to be correct,then the partial decay width could be as large as Γ(D +sJ (2317)→D +s π0)∼40keV,in agreement with the QCDSR calculation done inref.[16],where the meson D +sJ (2317)is considered as a ordinary c ¯s state.In Table II we show the partial decay width obtained by different theoretical groups.The first five calculationsassume a c ¯s picture for D +sJ (2317),while the last two assume a four-quark picture for it.Table II:The decay width Γ(D +sJ (2317)→D +s π0)from various theoretical approaches.ref.[13][15][20]Γ(keV)7±11610−1000|¯u u |0,which measures the isospin symmetry breaking in thequark condensate.Since,in our approach,the partial decay width is directly proportional to γ2,and since there is a large uncertainty in the value of γ,considering γin the range −2.6×10−2≤γ≤−2×10−3we get the partial decaywidth in the range 0.2keV ≤Γ(D +sJ (2317)→D +s π0)≤40keV.However,from other QCDSR calculation,we believethat the value of γis ∼−1×10−2,which gives the result shown in Eq.(16).As a final remark we would like to point out that if,instead of using a isoscalar current,we have used a isovector current for D sJ (2317)(as suggested in ref.[27]),the difference in Eq.(11)would be a factor 2in the place of γ.In this case the decay would be isospin allowed and the partial width would be ∼230MeV,much bigger than the experimental upper limit to the total width Γ∼5MeV.Acknowledgements[5]S.Godfrey and N.Isgur,Phys.Rev.D32,189(1985);S.Godfrey and R.Kokoshi,Phys.Rev.D43,1679(1991).[6]W.Bardeen,E.Eichten and C.Hill,Phys.Rev.D68,054024(2003).[7]Y.-B.Dai,C.-S.Huang,C.Liu and S.-L.Zhu,Phys.Rev.D68,114011(2003).[8]G.S.Bali,Phys.Rev.D68,071501(R)(2003).[9]A.Dougall,R.D.Kenway,C.M.Maynard and C.Mc-Neile,Phys.Lett.B569,41(2003).[10]A.Hayashigaki and K.Terasaki,hep-ph/0411285.[11]S.Narison,Phys.Lett.B605,319(2005).[12]A.Deandrea,G.Nardulli and A.Polosa,Phys.Rev.D68,097501(2003).[13]P.Colangelo and F.De Fazio,Phys.Lett.B570,180(2003).[14]S.Godfrey,Phys.Lett.B568,254(2003).[15]Fayyazuddin and Riazuddin,Phys.Rev.D69,114008(2004).[16]W.Wei,P.-Z.Huang and S.-L.Zhu,hep-ph/0510039.[17]T.Barnes,F.E.Close and H.J.Lipkin,Phys.Rev.D68,054006(2003).[18]A.P.Szczepaniak,Phys.Lett.B567,23(2003).[19]E.van Beveren and G.Rupp,Phys.Rev.Lett.91,012003(2003).[20]H.-Y.Cheng and W.-S.Hou,Phys.Lett.B566,193(2003).[21]K.Terasaki,Phys.Rev.D68,011501(R)(2003).[22]L.Maiani,F.Piccinini,A.D.Polosa,V.Riquer,Phys.Rev.D71,014028(2005).[23]M.E.Bracco et al.,Phys.Lett.B624,217(2005).[24]H.Kim and Y.Oh,hep-ph/0508251.[25]T.Browder,S.Pakvasa and A.A.Petrov,Phys.Lett.B578,365(2004).[26]U.Dmitrasinovic,Phys.Rev.D70,096011(2004);Phys.Rev.Lett.94,162002(2005).[27]A.Hayashigaki and K.Terasaki,hep-ph/0410393.[28]R.L.Jaffe and F.Wilczek,Phys.Rev.Lett.91,232003(2003).[29]M.A.Shifman,A.I.and Vainshtein and V.I.Zakharov,Nucl.Phys.B147,385(1979).[30]L.J.Reinders,H.Rubinstein and S.Yazaky,Phys.Rep.127,1(1985).[31]B.L.Ioffe,Nucl.Phys.B188,317(1981);B191,591(E)(1981).[32]S.Narison,Spectral Sum Rules,World Scientific Lecture Notes in Physics26.[33]M.E.Bracco,F.S.Navarra,M.Nielsen,Phys.Lett.B454,346(1999).[34]T.V.Brito,F.S.Navarra,M.Nielsen,M.E.Bracco,Phys.Lett.B608,69(2005).[35]M.E.Bracco et al.,Phys.Lett.B521,1(2001).[36]H.G.Dosch,E.M.Ferreira,F.S.Navarra and M.Nielsen,Phys.Rev.D65,114002(2002).[37]B.L.Ioffe and A.V.Smilga,Nucl.Phys.B232,109(1984).[38]M.Eidem´u ller et al.,Phys.Rev.D72,034003(2005).[39]J.Gasser,H.Leutwyler,Nucl.Phys..B250,465(1985).[40]X.Jin,M.Nielsen,J.Pasupathy,Phys.Rev.D51,3688(1995);H.Forkel,M.Nielsen,Phys.Rev.D55,1471(1997),andreferences therein.[41]H.-C.Kim,M.M.Musakhanov,M.Siddikov,hep-ph/0508211.[42]I.Bediaga,M.Nielsen,Phys.Rev.D68,036001(2003).。