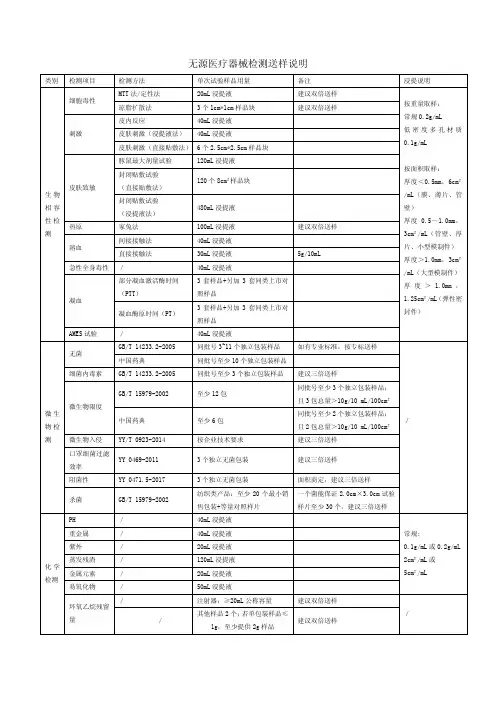
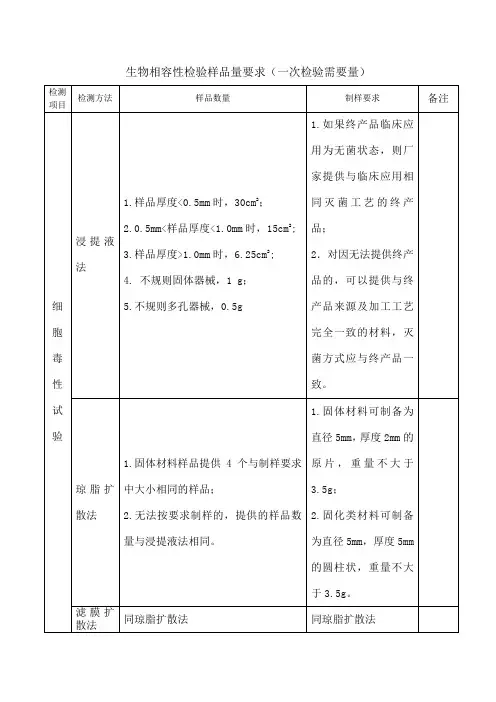
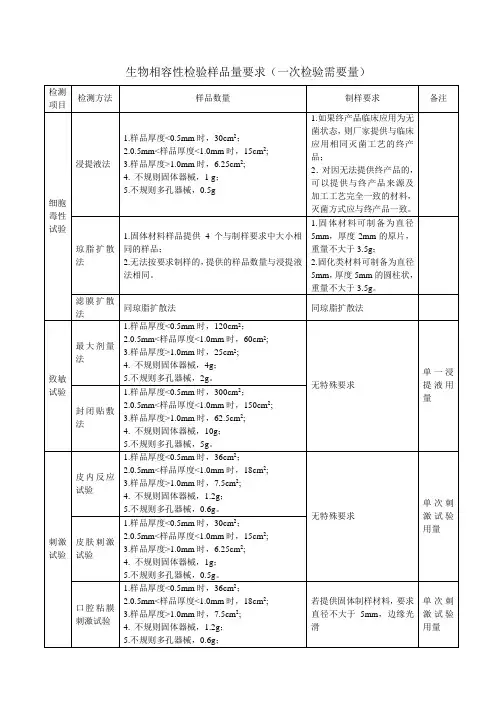
生物相容性检验样品量要求(一次检验需要量)
- 格式:doc
- 大小:89.51 KB
- 文档页数:4



生物样品分析方法的基本要求目录一、内容概述 (2)1.1 目的与意义 (3)1.2 生物样品分析的重要性 (3)二、生物样品的采集与保存 (4)2.1 采样原则与方法 (5)2.2 样品类型及采集量 (6)三、生物样品的前处理 (8)3.1 提取与分离 (9)3.2 净化与富集 (10)3.3 样品制备过程中的注意事项 (12)四、分析方法的建立与验证 (13)4.1 分析方法的选择 (15)4.2 方法学考察 (15)4.3 确证实验设计与实施 (17)五、生物样品分析质量控制 (18)5.1 质量保证与控制体系 (19)5.2 检测方法的性能指标 (21)5.3 质量控制图的绘制与应用 (22)六、生物样品分析结果的报告与解释 (24)6.1 结果报告的内容与格式 (25)6.2 结果的解释与判断 (27)6.3 异常值的分析与处理 (28)七、生物样品分析方法的应用与发展趋势 (30)7.1 应用领域与案例分析 (32)7.2 新技术、新方法的发展趋势 (34)一、内容概述生物样品分析方法的基本要求是针对生物样本进行科学研究的基础工具与技术方法的具体规范,主要涉及到实验设计、样本采集、样本处理以及分析方法等内容。
本文内容概述将概述该分析方法的整体框架与核心内容。
本文将阐述生物样品分析的基本目的和意义,强调其对于生物科学研究的重要性。
会介绍分析方法的实验设计部分,包括实验对象的选取原则、实验设计的合理性和科学性等要求。
本文将详细介绍样本采集的要求,包括采样点的选择、采样时间的选择以及采样技术的标准化等。
本文将重点介绍样品处理过程,包括样品的保存、预处理以及化学或物理性质的调整等步骤,以确保样品的完整性和准确性。
本文将强调分析方法的建立与优化,包括实验技术的选择与应用、数据分析与解读等关键环节,以确保分析结果的准确性和可靠性。
生物样品分析方法的基本要求涵盖了实验设计、样本采集、样本处理和分析方法等多个方面,旨在确保生物样品分析的准确性和可靠性,为科学研究提供可靠的实验数据支撑。

生物相容性测试和标准1.基本含义生物相容性指的是医用材料和病人的组织和生理系统间的相互适应性。
医用材料之所以能在临床取得成功并能安全使用,主要缘于其良好的生物相容性。
通常,一些医用材料在使用过程中会释放有毒物质,导致与病人不“相容”。
出于监控生物相容性的目的,一般会在坏的情况下模拟使用医用材料及其萃取物,从而确保在正常使用条件下的安全性。
ISO 10993 中,第1到第23部分规定了一系列强制标准来评价医用材料的生物相容性。
2.测试项目ISO10993标准通常包括的测试项目有体外细胞毒性测试、皮肤刺激性测试、致敏试验。
1)细胞毒性-- 是所有医疗器材都必须做的测试。
①主要参考标准:ISO 10993-5:2009医疗器械生物学评价第5部分:体外细胞毒性试验。
(国标使用的标准是GB/T 16886.5-2003)。
②仪器设备:高压灭菌器、恒温摇床、钢直尺、电子天平、CO2培养箱、倒置显微镜、超净工作台、酶联免疫检测仪等。
③实验浸提液:含10%胎牛血清的MEM浸提(阴性对照样品)④试验受体:小鼠成纤维细胞2)皮肤刺激-- 人体接触的医疗器材可能会释放造成皮肤、黏膜、或眼睛刺激反应的化学物质。
简单的说,这些刺激的现象是因为局部皮肤的发炎而造成红肿、甚至发热和疼痛的感觉。
许多的化学物质都能造成实时的或者是慢性的刺激反应。
这些化学物质包括医材的添加物、制程或生产的催化剂、或是不小心污染的物质。
譬如说:因环氧乙烷灭菌后所残留下的物质若没有降低到一定的程度就会造成人体的刺激反应?又如清洗医疗器材用的清洁液若没有适度的去除也会对使用者或病患造成意外的刺激反应。
①主要参考标准:ISO 10993-23:2021医疗器械生物学评价第23部分:刺激试验(国标使用的标准是GB/T 16886.10-2017)。
②仪器设备:电子秤、欧式大容量恒温培养振荡器、钢直尺、立式压力蒸汽灭菌器。
③实验浸提液:0.9%氯化钠注射液(阴性对照样品)④试验受体:新西兰兔3)致敏试验-- 过敏通常是因为重复或长期的与化学物质接触而引起系统的反应。

无菌检验样品用量(一次检验用量)
微生物样品用量(包含预留量)
抑菌试验送样要求
1.数量:见表格
2.提供对照样品,对照样品为:与样品相同材质,同等大小,但不含抗菌材料,
且经灭菌处理。
对照样品的数量同样品数量。
3.企标需明确试验菌种、作用时间及试验方法。
杀菌试验送样要求
1.数量:见表格
2.提供对照样品(与样品相同材质,同等大小,但不含抗菌材料,且经灭菌处理),
对照样品的数量同样品数量。
3.提供中和剂(在到达规定作用时间时能及时中止样品对微生物的杀灭作用,且
中和剂本身及其与样品的反应产物对微生物无抑制或杀灭作用,对培养基无不良影响)。
中和剂需无菌。
4.中和剂需进行鉴定,鉴定合格的中和剂方可用于杀菌试验。
如中和剂鉴定不合
格则该项试验不合格。
5.企标需明确试验菌种、作用时间及试验方法。

生物相容性试验项目
1.试验要求
1.1 细胞毒性:细胞毒性应不大于2级。
1.2 皮内反应:试验样品与溶剂对照综合平均记分之差应不大于1.0。
1.3 迟发型超敏反应:应无迟发型超敏反应。
2. 试验方法
2.1细胞毒性试验:
以细胞完全培养基为浸提介质,按照6cm2/mL的比例,(37±1)℃条件下浸
提24 h,根据GB/T 16886.5-2003中规定的浸提液法进行试验,结果应符合
1.1条的要求。
2.2 皮内反应试验:
分别以生理盐水和植物油为浸提介质,按照6cm2/mL的比例,(37±1)℃条
件下浸提(72±2)h,根据GB/T 16886.10-2005中规定的皮内反应方法进行
试验,结果应符合1.2的要求。
2.3迟发型超敏反应试验:
分别以生理盐水和植物油为浸提介质,按照6cm2/mL的比例,(37±1)℃条
件下浸提(72±2)h,根据GB/T 16886.10-2005中规定的最大剂量法进行试
验,结果应符合1.3的要求。
2015/3/12。

无菌医疗器械质量检验相关标准(一)医疗器械生物相容性试验GB/T 16886。
3 医疗器械生物学评价第 3部分:遗传毒性、致癌性和生殖毒性试验GB/T 16886.4 医疗器械生物学评价第 4部分:与血液相互作用试验选择GB/T 16886.5 医疗器械生物学评价第 5部分:体外细胞毒性试验GB/T 16886.6 医疗器械生物学评价第 6部分:植入后局部反应试验GB/T 16886。
7 医疗器械生物学评价第 7部分:环氧乙烷灭菌残留量GB/T 16886.8 医疗器械生物学评价第 8部分:生物学试验参照样品的选择和定性GB/T 16886.10 医疗器械生物学评价第 10 部分:刺激与致敏试验GB/T 16886.11 医疗器械生物学评价第 11 部分:全身毒性试验GB/T 16886。
12 医疗器械生物学评价第 12 部分:样品制备和参照样品GB/T 16886.13 医疗器械生物学评价第 13 部分:聚合物医疗器械降解产物的定性与定量GB/T 16886。
14 医疗器械生物学评价第 14 部分:陶瓷降解产物的定性与定量分析GB/T 16886。
15 医疗器械生物学评价第 15 部分:金属与合金降解产物的定性与定量GB/T 16886。
16 医疗器械生物学评价第 16 部分:降解产物与可溶出物的毒代动力学研究设计)GB/T 16886.17 医疗器械生物学评价第 17 部分:根据健康风险评价确定可溶出物允许极限的方法GB/T 16886.18 医疗器械生物学评价第 18 部分:材料化学定性GB/T 16886.19 医疗器械生物学评价第 19 部分:材料理化、机械和形态定性GB/T 16886。
20 医疗器械生物学评价第 20 部分:医疗器械免疫毒理学试验原理和方法GB/T16175 医用有机硅材料生物学评价试验方法GB/T 16886.9 医疗器械生物学评价第 9部分:潜在降解产物的定性与定量总则YY/T 0473 外科植入物聚交酯共聚物和共混物体外降解试验YY/T 0474 外科植入物用聚L-丙交酯树脂及制品体外降解试验YY/T 0567。
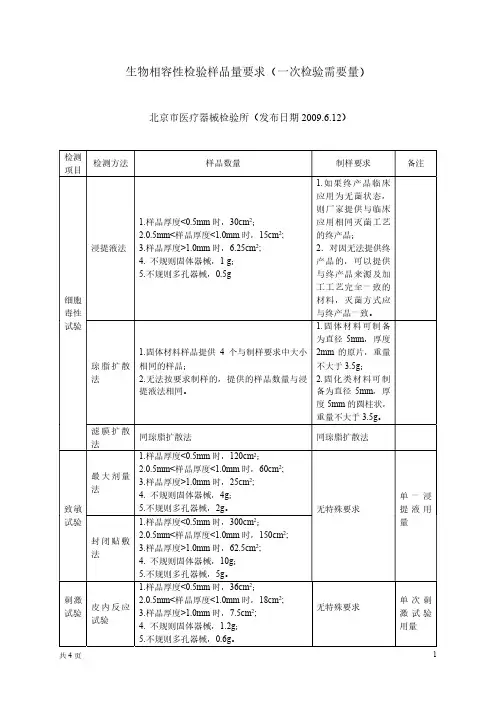
生物相容性检验要求及试验方法编写示例为规范实施GB/T 16886系列等生物相容性评价标准,方便注册产品标准相关章节的编写,我所生物相容性检验室编写了生物相容性部分检验项目要求及试验方法的示例,供相关人员参考。
生物相容性试验要求常用试验项目:1.细胞毒性:细胞相对增殖率应不小于70%。
(采用GB/T16886.5-2017中MTT法时)细胞毒性应不大于X级。
(琼脂扩散法等的要求,企业根据自身产品性质和风险确定其级别的要求)2.皮内反应:试验样品与溶剂对照综合平均记分之差应不大于1.0。
3.迟发型超敏反应:应无迟发型超敏反应。
其它试验项目刺激试验分类:4.原发性皮肤刺激:原发性皮肤刺激指数应不大于0.4。
5.阴道刺激:刺激指数应不大于4(极轻)/8(轻度)。
(企业根据自身产品性质和风险进行要求)6.口腔黏膜刺激:刺激指数应不大于4(极轻)/8(轻度)。
(企业根据自身产品性质和风险进行要求)7. 眼刺激:试验样品不引起眼刺激反应。
全身毒性试验分类:8. 急性全身毒性:应无急性全身毒性反应。
9. 亚急性全身毒性:应无亚急性全身毒性反应。
10. 亚慢性全身毒性:应无亚慢性全身毒性反应。
11. 热原:试验样品应无热原反应。
遗传毒性试验:12. 鼠伤寒沙门氏菌回复突变试验(Ames试验):诱变应为阴性。
13. 小鼠淋巴瘤细胞基因突变试验:诱变应为阴性。
14. 染色体畸变试验:诱变应为阴性。
15.植入试验:与对照样品相比,应不引起组织学反应或仅引起轻微组织学反应。
(注:植入试验应明确植入部位和周期,如:12周肌肉植入试验)生物相容性试验方法:原则:主要明确浸提介质、浸提比例、浸提温度、浸提时间和所用方法1.细胞毒性:以细胞完全培养基为浸提介质,按照0.2g/mL的比例,(37±1)℃条件下浸提24 h,根据GB/T 16886.5-2017中规定的浸提液法进行试验,结果应符合X.X的要求。
注:浸提比例请企业根据样品特点按照GB/T 16886.12中相应条件进行选择;同时根据所选择的细胞毒性不同试验方法(如浸提液法MTT、琼脂扩散法、直接接触法等)在方法中进行说明。
1 目的规范微生物限度检验操作,确保检验结果准确、可靠2 依据《中国药典》 2015 版。
3 范围本标准适用于微生物限度检验。
4 责任中心化验室负责人:对本规程的实施负责。
中心化验室检验人员:严格按照本规程执行检验操作5 程序5.执行标准:《中国药典》 2015 版。
15.抽样:照《取样标准操作规程》进行26 内容6.需氧菌总数、霉菌和酵母菌总数计数1计数方法包括平皿法、薄膜过滤法和最可能数法(MPN法)。
检查时,按已验证的计数方法进行供试品的需氧菌总数、霉菌和酵母菌总数的测定。
6.1.1 设备、仪器及用具6.1.1.1 设备微生物限度检测室、净化工作台、生化培养箱(30〜35°C)、生化培养箱(20〜25°C)、电热恒温水浴锅、烘箱、脉动真空灭菌柜、紫外灯等。
6.1.1.2 仪器及器皿显微镜、托盘天平、锥形瓶、研钵、培养皿、量筒、试管及塞、移液枪及吸头、薄膜过滤器等。
6.1.1.3 用具大、小橡皮乳头,洁净工作服、口罩、医用手套、接种环、酒精灯、酒精棉球、灭菌剪刀、灭菌镊子、不锈钢药匙、试管架、火柴、记号笔、薄膜过滤膜等。
6.1.2 试液6.1.2.1 消毒液A .0.1% 新洁尔灭溶液B. 75聽醇溶液6.1.2.2 稀释液、试剂及配制A. pH7.0无菌氯化钠-蛋白胨缓冲液:取磷酸二氢钾 3.56g、无水磷酸氢二钠5.77g、氯化钠4.3g、蛋白胨1.0g,加纯化水1000ml,微温溶解,滤清,分装,121°C灭菌15分钟。
B. 聚山梨酯80C. 无菌十四烷酸异丙酯D. pH6.8无菌磷酸盐缓冲液6.1.3 培养基胰酪大豆胨琼脂培养基、胰酪大豆胨液体培养基、沙氏葡萄糖琼脂培养基6.1.4 操作方法6.1.4.1 试验前准备6.1.4.1.1 将供试品及所有已灭菌的平皿、锥形瓶、试管、移液枪吸头( 1ml、 10ml)、量筒、稀释液、培养基等移至传递窗内。
每次试验所用物品必须事先计划,准备足够用量,避免操作中出入操作间。
进行生物学评价试验需提供的材料清单一、委托协议说明:1. 委托协议中应注明进行试验项目选择的实验对象是最终产品、取自最终产品中有代表性的样品或与最终产品以相同的工艺过程制得的材料。
2. 请根据产品特点和临床使用特性,按照GB/T16886.12《医疗器械生物学评价第12部分:样品制备与参照样品》的规定选择试验液制备方法,确定浸提介质、比例、温度和时间,或依据常用操作并在论证的基础上提供一个标准化的方法,在多数情况下产品应使用适宜的加严条件。
若受试样品不适合用二甲基亚砜等有机溶剂做为浸提介质,可采用其他浸提介质,如遗传毒性可改用细胞培养基等。
3. 若样品制备中有其他特殊要求,如需完整浸提等应特殊注明。
如需对浸提液调节PH值或过滤、离心等方法去除悬浮的粒子,请在委托声明中写出关键步骤(如调节PH的介质,过滤的滤膜孔径,离心速率等);如样品存在吸胀、固化等,请注明相应处理的方式、吸胀量(按不同的浸提介质分别提供)、比例及条件等相关信息。
4. 如果不同的组件需要分别进行试验,委托协议中应分别进行规定。
5. 细胞毒性:如无特殊规定,本中心采用ISO10993.5-2009规定的细胞毒性试验定量法(MTT法)进行细胞毒性试验。
6. 刺激试验:下列的委托协议范例中只列出了皮内反应和皮肤刺激,GB/T16886.10中规定了多种刺激试验检测方法,请根据医疗器械的预期用途和使用部位选择适宜的检测方法。
7.皮肤刺激试验:GB/T16886.10-2005中6.3动物皮肤刺激试验规定了单次接触试验和多次接触试验。
委托方应根据自身产品的情况选择。
国家标准中规定单次接触试验:单次接触试验样品至少4h后,再除去敷贴片。
分别在除去敷贴片后1h、24h、48h和72h记录各接触部位情况。
如存在持久性损伤则有必要延长观察时间,以评价这种损伤的可逆性或不可逆性,但延长期不超过14d。
多次接触试验:多次接触试验应仅在急性单次接触试验完成后进行(至少在观察72h后)。