第12章 羧酸
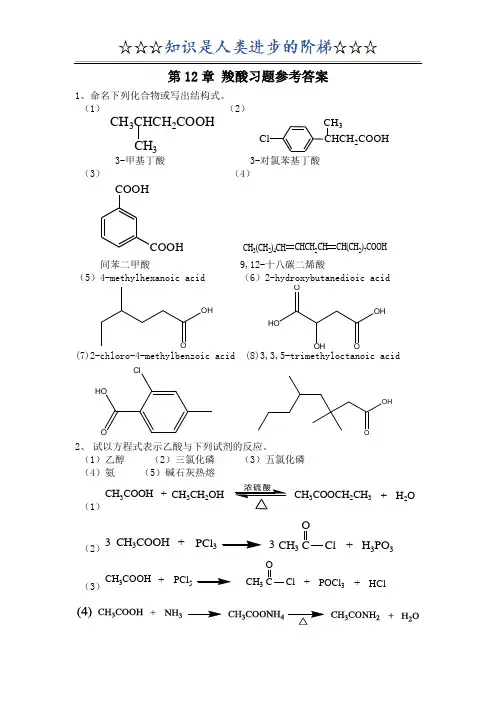
- 格式:docx
- 大小:862.04 KB
- 文档页数:15
第12章 羧酸
§12.1 羧酸的分类、结构与命名
12.1.1 结构和分类
1、定义
分子中含有基团(羧基)的有机化合物称为羧酸。
2、羧酸的结构通式:R-COOH (-R=烷基或芳基)
3、羧酸(RCOOH)的分类方法
按照羧基连的烃基构造: 按照分子中羧基的数目:
脂肪族羧酸(饱和及不饱和) 一元羧酸
脂环族羧酸 二元羧酸
芳香族羧酸 多元羧酸
其中链状的一元羧酸(包括饱和的及不饱和的)通称为脂肪酸
12.1.2 命名
1、系统命名法
A、饱和脂肪酸的命名
1)选择含有羧基的最长碳链为主链,并按主链碳数称“某酸”;
2)从羧基碳原子开始编号,用阿拉伯数字标明取代基的位置;
3)并将取代基的位次、数目、名称写于酸名前。
如:
B、不饱和脂肪酸的命名
1)选择包括羧基碳原子和各C=C键的碳原子都在内的最长碳链为主链,根据主链上碳原子的数目称“某酸”或“某烯(炔)酸”;
2)从羧基碳原子开始编号;
3)在“某烯(炔)酸”前并注明取代基情况及双键的位置。
如:
2, 4–二甲基–3–戊烯酸 (E) –丁烯二酸 3-苯基丙烯酸 COHOC H 3 C H 2 C H 2 C O O H 丁酸
C H 3 C H
C H 3 C H
C H 3 C H 2 C O O H 3 , 4 - 二甲基戊酸
- 二甲基戊酸 1 2 3 4
5
CH3CCHCHCH3CH3COOHCCCOOHHOOCHHC、脂环族羧酸的命名
1)羧基直接连在脂环上时,可在脂环烃的名称后加上“羧酸或二羧酸”等词尾;
2)不论羧基直接连在脂环上还是在脂环侧链上,均可把脂环作为取代基来命名。
如:
D、芳香族羧酸的命名
1)以芳甲酸为母体;
2)若芳环上连有取代基,则从羧基所连的碳原子开始编号,并使取代基的位次最小。
如:
E、二元酸的命名
选包括两个羧基碳原子在内的最长碳链作为主链,按主链的碳原子数称为“某二酸”。
如:
2、普通命名法(适用于简单结构羧酸)
1)选含有羧基的最长的碳链为主链;
2)取代基的位置从羧基邻接的碳原子开始,用希腊字母表示,依次为α,β,γ,δ,ε等,最末端碳原子可用ω表示。
如:
§12.2 羧酸的物理性质
1、物态:C1~C3刺激臭味液体; C4~C9腐败气味油状液体;
C10以上羧酸为固体。
2、溶解度: 注意:羧基在芳烃侧链上,以脂肪酸为母体! 羧酸具有比较广泛的溶解性:
甲酸至丁酸能与水混溶,从戊酸开始随相对分子质量增加,水溶性迅速降低,癸酸以上的羧酸不溶于水;
低级的饱和二元羧酸也可溶于水,并随碳链的增长而溶解度降低;
芳酸的水溶性极微,常常在水中重结晶;
脂肪族一元羧酸一般都能溶于乙醇、乙醚、卤仿等有机溶剂中。
3、沸点和熔点
由于羧酸分子之间能由两个氢键互相结合形成双分子缔合二聚体,羧酸的沸点和熔点比相同分子量的烷烃及其他极性分子的沸点与熔点都高。
饱和一元羧酸的沸点随相对分子量的增加而升高;
碳数相同的直链一元羧酸的沸点比支链的高;
羧酸的熔点随着碳原子数的增加而呈锯齿状上升。
偶数碳羧酸的m.p高于相邻两奇数碳羧酸的m.p
『原因:奇数碳羧酸的分子对称性低结构不紧凑。』
§12.3 羧酸的制备方法
Ⅰ、氧化反应 Ⅱ、水解反应 Ⅲ、羧化反应(插入CO2)
①高级脂肪烃氧化 ①腈的水解 ①格氏试剂与CO2反应
②烯烃、炔烃的氧化断裂 ②油脂的水解 ②苯酚钠/钾盐与CO2反应(酚酸反应)
③芳烃的侧链氧化
④1o醇、醛的氧化
⑤甲基酮的卤仿反应
12.3.1 氧化反应
1、高级脂肪烃氧化
RCH2CH2RˊRCOOH+ RˊCOOH
2、烯烃、炔烃的氧化断裂 RCH=CHRˊRCOOH+ RˊCOOH
RC≡CRˊRCOOH+ RˊCOOH
3、芳烃的侧链氧化 22O,MnO+4KMnO,H22OHO 4、1o醇、醛的氧化
RCH2OHRCOOH
5、甲基酮的卤仿反应
12.3.2 水解反应
1、腈的水解
2、油脂的水解
12.3.3 羧化反应
1、格氏试剂与CO2反应
2、苯酚钠/钾盐与CO2反应(酚酸反应) ——Kolbe-Schmitt反应
邻位
对位
§12.4 羧酸的化学性质
1、羧基的结构特征
1)羧基碳原子为sp2杂化;
2)羧基中含处在同一平面的3个σ键;
3)碳p轨道与羰基氧原子的p轨道平行相互重叠形成一个π键;
4)羟基氧原子的未共用电子对与羰基上的π键形成pπ共轭。
+4+227KMnO,H, KCrO,HOrRCHO+32+31)AgNH2)HORCOOH产物比RX多一个C,与RCN不同 表明:羰基不是碳氧双键,而是介于单双键之间;同样羰基碳与羟基氧的C—O键也不是单键,而是介于单双键之间。
由于羧基中pπ共轭体系的存在,使得羧基不是羰基和羟基的简单加合,而是两者相互影响的统一体。
羟基氧上的电子云密度降低,O—H键变弱,容易断裂,电离出质子(H+),表现出一定的酸性(酸性比醇大);
羰基与亲核试剂的反应活性降低。不能再与HCN及含氮的亲核试剂进行加成反应,与醛酮的性质不同。
2、羧酸主要化学反应发生的位置
①O-H键易断裂表现出酸性
②-OH被取代的反应(实质是羰基的亲核加成-消除反应)
③羰基的亲核加成反应
④C-C键断裂发生脱羧反应
⑤α-H的取代反应
12.4.1 羧酸的酸性
1、羧酸的酸性
羧酸根负离子的p-π共轭效应,使氧上带的负电荷被平均分散在它的两个氧原子上,使离子比较稳定并且两个碳氧键是等同的。
有机化合物 羧酸 碳酸 苯酚 醇
pKa
3.5~5 6.38 10.0
15.9
羧酸属于弱酸,但比碳酸的酸性要强些。所以:
①羧酸可与Na2CO3或NaHCO3溶液发生反应,可用作鉴定酚和羧酸: RCOOH+NaHCO3→RCOONa+CO2+H2O
②加入无机强酸又可以使盐重新变为羧酸游离出来,此性质可利用来分离羧酸与不溶于水的或易挥发的物质:
RCOONa+HCl→RCOOH+NaCl 2、影响羧酸酸性的因素
由上式通式可知,削弱O-H键或使羧基负离子稳定的因素都会增强羧酸的酸性。
(1)电子效应 HOORH2O+ROO-H3O++ 21RCO-ORCOO-RCOO----21HOOH2O+OO-H3O++GG酸性; 0apK
A、脂肪酸
➢ 吸电子基团G有利于羧基负离子上的负电荷的进一步分散稳定性↗使酸性↗;
➢ 推电子基团G使其负电荷相应集中吸引质子的能力↗导致稳定性↘酸性↘。
推电子基团:
吸电子基团:
如:
1)由于推电子诱导效应使酸性减弱
CH3COOH CH3CH2COOH (CH3)2CHCOOH (CH3)3CCOOH
4.76 4.86 4.87 5.02
2)羧酸分子中引入的取代原子电负性愈强,吸电子诱导效应愈强,酸性愈强。
FCH2COOH ClCH2COOH BrCH2COOH ICH2COOH
2.67 2.87 2.90 3.16
3)羧酸分子中引入的吸(推)电子基团的数目愈多(加合性),吸电子诱导效应愈强(弱),酸性也愈强(弱);
CH3COOH ClCH2COOH Cl2CHCOOH Cl3CCOOH
4.76 2.87 1.36 0.36
4)吸电子基团距羧基的位置愈近,对羧基的影响愈大,酸性愈强
CH3CH2CHCOOH CH3CHCH2COOH CH2CH2CH2COOH
| | |
Cl Cl Cl
2.86 4.06 4.52
B、芳香酸
苯基具有吸电诱导效应和推电共轭效应,且推电共轭效应大于吸电诱导效应,因此苯基对羧基有推电子能力。
➢ 邻位时,除氨基外,都使苯甲酸的酸性增强;
➢ 间位时,吸电子基团使酸性增强。推电子基团则相反;
➢ 对位时,取代基为第一类定位基(除了F、Cl、Br、I外)使酸性减弱,其余的包括第二类定位基使酸性增强。 apKapKapKapK C、基团的场效应
具有强吸电子作用的邻位取代基(如F,NO2),由于它可在空间上对羧酸根施加空间诱导作用(通称场效应),使羧酸根上的负电荷通过空间场直接分散到邻位的吸电基上,结果使羧酸根的稳定性增加,因此使该取代酸的酸性比其间位和对位异构体的强。
(2)基团的立体效应
邻位的CH3、C2H5由于空间的拥挤,取代基破坏了羧基与苯环的共平面性,苯环对羧基的+C效应减弱甚至消失,使其酸性接近甲酸,这种立体效应使酸性比间位或对位取代的苯甲酸强。
(3)氢键的形成
邻羟基苯甲酸(水杨酸),由于羧酸根负离子与相邻的羟基可以通过形成氢键而使其稳定性增强,所以邻羟基苯甲酸的酸性也比其间位和对位异构体都强。
3、二元酸的酸性
二元酸分子中含有两个羧基,可以分两步离解:
酸性大小规律为:
二元酸pKa2>pKa1
二元酸pKa1
12.4.2 羧基中O-H键的反应
1、与碱反应成盐
2、羧酸负离子的亲核反应
羧酸钠盐是一弱的亲核试剂,可与活泼的卤代烃如苯甲基氯发生反应形成酯,也可在催化剂如四丁基铵盐作用下进行亲核取代反应。
3、活泼H与金属有机化合物反应
A、与RMgX反应——生成相应的烃
B、与RLi反应——制备酮 COOH(CH2)nCOOH(CH2)nCOOHCOO-(CH2)nCOO-COO-+ H+K1