国家药学申报资料格式
- 格式:pdf
- 大小:137.61 KB
- 文档页数:8





cde申报资料模板
以下是一些关于CDE申报资料的信息和注意事项:
CDE是药品审评中心的简称,是国家药品监督管理局下属的一个机构,负责药品审评和注册等工作。
在向CDE提交药品注册申请时,需要按照规定的格式和要求提交申报资料,以确保申请能够顺利通过审评。
申报资料的格式和要求因申请类型和药品种类的不同而有所不同,但一般包括以下内容:
1. 药品注册申请表:包括申请人信息、药品基本信息、研究情况、临床试验情况等。
2. 药学研究资料:包括药学研究总结、药学研究报告、质量标准、工艺研究资料、稳定性研究资料等。
3. 临床研究资料:包括临床试验总结、临床试验报告、伦理审查报告等。
4. 综述资料:包括药品概述、药学研究综述、临床研究综述、国内外文献综述等。
5. 其他资料:包括申请人资质证明文件、药品制造和质量控制证书等。
在准备申报资料时,需要注意以下几点:
1. 确保资料完整、真实、准确,符合规定的格式和要求。
2. 按照规定的顺序和要求整理资料,确保审评人员能够顺利查阅和理解。
3. 重视药学研究和临床研究的科学性和规范性,确保数据真实可靠。
4. 注意保护知识产权,避免侵犯他人专利或商标。
5. 在提交申请前,进行自查和审核,确保没有遗漏或错误。
如果您需要更具体的帮助,建议您查阅CDE官方网站或咨询专业人士。

药品注册分类:化学药品六类注册申请分类:仿制药品注册申请药品名称:苯磺酸氨氯地平片(XX g、XX g)资料项目名称:药学研究CTD格式申报资料研究机构名称:XXX制药有限公司研究机构地址:XXXXXXXXX研究机构主要研究者:XXX研究机构电话:XXX注册申请联系人姓名:XXX原始资料的保存地点:XXX制药有限公司注册申请机构联系电话:XXXXXXXXX药品注册申请人:XXX制药有限公司苯磺酸氨氯地平片申报资料(药学部分)目录3.2.P.1 剂型及产品组成 (3)3.2.P.2 产品开发 (4)3.2.P.2.1 处方组成 (4)3.2.P.2.1.1 原料药 (4)3.2.P.2.1.2 辅料 (5)3.2.P.2.2 制剂研究 (5)3.2.P.2.2.1 处方开发过程 (5)3.2.P.2.3 生产工艺的开发 (20)3.2.P.2.4 包装材料/容器 (23)2.3.P.2.5 相容性 (23)3.2.P.3 生产 (23)3.2.P.3.1生产商 (23)3.2.P.3.2批处方 (23)3.2.P.3.3 生产工艺和工艺控制 (24)3.2.P.3.4 关键步骤和中间体的控制 (25)3.2.P.3.5 工艺验证和评价 (26)3.2.P.4 原辅料的控制 (31)3.2.P.5 制剂的质量控制 (31)3.2.P.5.1质量标准 (31)3.2.P.5.2 分析方法 (31)3.2.P.5.3 分析方法的验证 (34)3.2.P.5.4 批检验报告 (93)3.2.P.5.5 杂质分析 (97)3.2.P.6 对照品 (100)3.2.P.7 稳定性 (101)3.2.P.7.1稳定性总结 (101)3.2.P.7.2上市后的稳定性承诺和稳定性方案 (102)3.2.P.7.3 稳定性数据 (102)申报资料正文3.2.P.1 剂型及产品组成苯磺酸氨氯地平片是一种独特的具有高度血管选择性的长效二氢吡啶类钙离子拮抗剂,是心血管治疗药物中比较理想的长效降压药,也是近几年来世界处方量最大的高血压和心绞痛治疗药物。

药学专业技术资格的评审申报表填写范本一、申报人基本情况
姓名:
性别:
出生年月:
身份证号码:
学历:
专业:
毕业院校:
参加工作时间:
所在单位:
联系方式:
二、申报的资格级别
(请在下面选择符合自己条件的资格级别,可以选择多个)•[ ] 初级药学工程师
•[ ] 中级药学工程师
•[ ] 高级药学工程师
•[ ] 初级药品注册师
•[ ] 中级药品注册师
•[ ] 高级药品注册师
•[ ] 初级药剂师
•[ ] 中级药剂师
•[ ] 高级药剂师
三、申报审核材料清单
•[ ] 申请书
•[ ] 身份证明
•[ ] 学历证书
•[ ] 工作证明
•[ ] 荣誉证书
•[ ] 专业技术资格证书
四、个人简介
请从以下几个方面介绍自己的工作经历和专业技术水平(字数不少于300字):
1. 个人工作经历
(请列举自己的工作经历,包括工作岗位、工作时间、主要工作内容等,字数
不少于200字)
2. 个人专业技术水平
(请简要介绍自己的专业技术水平,包括曾获得的荣誉,参加的重要项目、研
究成果等,字数不少于100字)
五、申报人承诺
本人承诺所填写的申报材料真实有效,如有不实之处,愿承担一切责任。
申报人签名:
日期:
小结
以上是药学专业技术资格的评审申报表填写范本,申报人可以根据自己的情况
进行相应的填写。
在填写申报表时,需注意表格格式的规范和字数要求。
祝愿所有申报人顺利通过专业技术资格评审。

药品注册申报的体例与规范文档编制序号:[KK8UY-LL9IO69-TTO6M3-MTOL89-FTT688]药品注册申报资料的体例与整理规范为加强药品注册纸质申报资料的规范管理,特制定本规范。
当申报资料同时进行CTD格式提交时,纸质申报资料的体例设置必须与CTD申报格式电子文档相一致。
1.申报资料的体例要求字体、字号、字体颜色、行间距离及页边距离字体中文:宋体英文:Times New Roman字号中文:不小于小4号字,表格不小于5号字;申报资料封面加粗4号;申报资料目录小4号,脚注5号字。
英文:不小于12号字。
字体颜色黑色行间距离及页边距离行间距离:单倍。
纵向页面:左边距离不小于厘米、上边距离不小于2厘米、其他边距不小于1厘米。
横向页面:上边距离不小于厘米、右边距离不小于2厘米、其他边距不小于1厘米。
页眉和页脚:信息在上述页边距内显示,保证文本在打印或装订中不丢失信息。
纸张规格申报资料使用国际标准A4型(297mm×210mm)规格、纸张重量80g,纸张全套双面或全套单面打印,内容应完整、清楚,不得涂改。
纸张性能申报资料文件材料的载体和书写材料应符合耐久性要求。
加盖印章除《药品注册申请表》、相关受理文件及检验机构出具的检验报告外,申报资料应逐个封面加盖申请人印章(多个申请人联合申报的,应加盖所有申请人印章),封面与骑缝处加盖临床研究基地有效公章,封面印章应加盖在文字处。
加盖的印章应符合国家有关用章规定,并具法律效力。
2.申报资料的整理要求申报资料封面申报资料袋封面档案袋封面注明:申请分类、注册分类、药品名称、本袋所属第X套第X袋每套共X袋、原件/复印件联系人、联系电话、申请单位名称。
申报资料袋封面(档案袋)应采用国家局统一格式(条码信息)的封面。
多规格的品种为同一册申报资料时,申报资料袋封面,需显示多规格的条形码的受理号(同一封面)。
申报资料项目封面每项资料加“封面”,每项资料封面上注明:药品名称、资料项目编号、项目名称、申请机构、联系人姓名、电话、地址。

cde申报资料模板CDE(China Drug Evaluation)申报资料是指在中国进行药物注册申报时所需要提交的相关材料。
这些资料主要用于对药物的安全性、有效性和质量进行评价,并最终决定是否批准该药物上市。
为了帮助申报人员准备相应的申报资料,以下是一份相关参考内容,供申报人员参考。
一、企业资质文件1.1 企业营业执照副本复印件1.2 企业生产许可证副本复印件1.3 企业药品生产许可证副本复印件1.4 企业药品GMP证书复印件二、研究开发资料2.1 药物研发计划:包括研究目标、研究设计、研究方案等相关信息2.2 药物临床试验资料:包括临床试验计划、试验结果、药物临床安全性评估等信息2.3 临床试验伦理委员会批准文件及委员会评估报告2.4 药物质量控制文件:包括药物的质量标准和质量控制方法等信息2.5 药物不良反应监测和报告文件:包括不良反应的监测和报告方法、不良反应事件的报告记录等信息三、药物监管文件3.1 药物批准文件:包括国内外相关药物批准文件的复印件3.2 国内外相关药物市场情况的调查分析报告四、药物学资料4.1 药物化学和配方学资料:包括药物的化学结构、化学性质以及配方的成分和比例等信息4.2 药物制剂学资料:包括药物的制剂形式、质量控制和稳定性等信息4.3 药物药理学资料:包括药物的作用机制、药效学、药动学以及药物之间的相互作用等信息4.4 药物毒理学资料:包括药物的急性毒性、亚急性毒性、慢性毒性以及生殖毒性等信息五、药物临床试验资料5.1 药物临床试验同意书和知情同意书5.2 药物临床试验方案和研究计划5.3 药物临床试验结果的统计分析报告5.4 药物临床试验的不良反应监测和报告文件六、药物充分性和适应性资料6.1 药物充分性资料:包括药物的临床试验结果、药物安全性和有效性的评价等信息6.2 药物适应性资料:包括药物的适应症、用法和用量等信息以上是CDE申报资料模板的相关参考内容,具体的申报要求还需要根据相关法规和CDE的相关通知进行调整。

药学m4申报资料药学M4申报资料一、个人简介我是XXX,性别男,出生于XXXX年XX月XX日,籍贯XXXXX。
我于XXXX年参加高考并考入XXXXX大学药学专业。
大学四年的学习期间,我通过系统而全面的课程学习,掌握了药学领域的基础知识和技能,并取得了优秀的学业成绩。
二、学术背景在大学期间,我以药学为主攻方向,修读了药物化学、药剂学、药理学、药用植物学等相关专业课程,并且在期间积极参与了药学实验室的科研工作。
此外,我也参与了多项药学类研究项目,积累了丰富的实践经验。
三、研究经历在大学期间,我参与了多项科研项目,主要包括:1. XXXX项目:该项目主要研究了某种药物的合成及其抗癌活性。
我负责了该项目的药物合成和纯化工作,并且在该过程中积累了一定的化学合成技能和实验操作经验。
2. XXXX项目:该项目涉及到抗生素的药物研发和预防耐药性的研究。
我主要负责了药理学方面的实验设计和数据分析工作,并且为项目取得了一定的研究成果。
此外,我还参与了一系列与药学相关的学术论文的撰写,并有多篇论文在国内核心期刊上发表。
四、实践经历在大学期间,我也积极参与了一些与药学相关的实践活动,主要包括实习和社会工作。
具体如下:1. 实习经历:我先后在X天晴药业有限公司和X医院进行了为期数周的实习。
在天晴药业有限公司,我主要参与了生产和质量控制的工作,熟悉了药品生产和质检过程,并且积累了一定的临床用药知识。
在X医院,我主要负责了药学咨询和药品管理工作,锻炼了临床药学实践技能。
2. 社会工作经历:我还积极参与了一系列药学类的社会工作,包括药品宣传、药物安全教育等。
通过这些社会工作,我提高了自己的沟通能力和团队协作能力。
五、荣誉奖项在大学期间,我取得了一系列的荣誉奖项,包括:1. 优秀学生:在大学四年中,我荣获了优秀学生奖学金三次,并被评为优秀共青团员。
2. 科技竞赛:我参加了多个省级和校级的科技竞赛,并荣获多项奖项,包括XXX竞赛一等奖等。
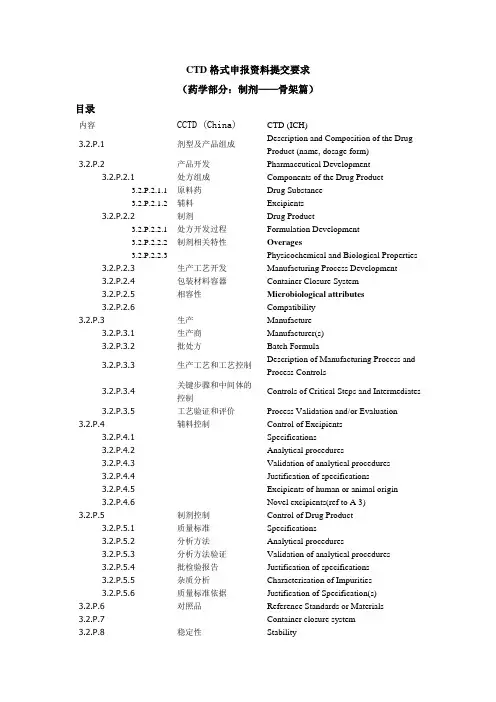
CTD格式申报资料提交要求(药学部分:制剂——骨架篇)目录内容CCTD (China) CTD (ICH)3.2.P.1 剂型及产品组成Description and Composition of the Drug Product (name, dosage form)3.2.P.2 产品开发Pharmaceutical Development3.2.P.2.1 处方组成Components of the Drug Product3.2.P.2.1.1 原料药Drug Substance3.2.P.2.1.2 辅料Excipients3.2.P.2.2 制剂Drug Product3.2.P.2.2.1 处方开发过程Formulation Development3.2.P.2.2.2 制剂相关特性Overages3.2.P.2.2.3 Physicochemical and Biological Properties3.2.P.2.3 生产工艺开发Manufacturing Process Development3.2.P.2.4 包装材料容器Container Closure System3.2.P.2.5 相容性Microbiological attributes3.2.P.2.6 Compatibility3.2.P.3 生产Manufacture3.2.P.3.1 生产商Manufacturer(s)3.2.P.3.2 批处方Batch Formula3.2.P.3.3 生产工艺和工艺控制Description of Manufacturing Process and Process Controls3.2.P.3.4 关键步骤和中间体的控制Controls of Critical Steps and Intermediates3.2.P.3.5 工艺验证和评价Process Validation and/or Evaluation 3.2.P.4 辅料控制Control of Excipients3.2.P.4.1 Specifications3.2.P.4.2 Analytical procedures3.2.P.4.3 Validation of analytical procedures3.2.P.4.4 Justification of specifications3.2.P.4.5 Excipients of human or animal origin3.2.P.4.6 Novel excipients(ref to A 3)3.2.P.5 制剂控制Control of Drug Product3.2.P.5.1 质量标准Specifications3.2.P.5.2 分析方法Analytical procedures3.2.P.5.3 分析方法验证Validation of analytical procedures3.2.P.5.4 批检验报告Justification of specifications3.2.P.5.5 杂质分析Characterisation of Impurities3.2.P.5.6 质量标准依据Justification of Specification(s)3.2.P.6 对照品Reference Standards or Materials 3.2.P.7 Container closure system3.2.P.8 稳定性Stability3.2.P.8.1 稳定性总结 Stability Summary and Conclusion 3.2.P.8.2 上市后稳定性研究方案和承诺 Post-approval Stability Protocol and Stability3.2.P.8.3稳定性数据Stability Data个人定义CTD :ICH 的要求,英文水平所限,仅罗列上,没有核对。
新药Ⅱ期和Ⅲ期临床试验药学申报资料的内容及格式要求目录Ⅰ. 前言 (1)Ⅱ. 背景 (1)现行法规要求A. (1)B.一般原则 (2)Ⅲ. Ⅱ期临床研究 (4)A. (4)原料药制剂B. (7)Ⅳ. Ⅲ期临床研究 (9)原料药A. (10)制剂B. (13)Ⅴ. 安慰剂 (16)Ⅵ. 标签 (16)Ⅶ. 环境评估 (16)新药Ⅱ期和Ⅲ期临床试验药学申报资料的内容及格式要求Ⅰ. 前言本指导原则为进行新药临床研究申请的申办者,提供新药临床研究申请Ⅱ、Ⅲ期临床研究申报的药学资料方面的建议。
本指导原则适用于人用化学药物,不适用于植物药、天然物质衍生获得的药物或使用生物技术产生的蛋白质类药物或其它生物制品。
本指导原则的目的是:(1)确保充足的数据提交给监管机构,从药学角度评价拟进行的临床研究的安全性和质量;(2)通过阐明Ⅱ、Ⅲ期临床试验中药学资料的类型、范围和报告格式,加快新药上市;(3)推动协调药物研发。
根据临床研究所处阶段、拟定的人体试验及资料是否与安全性有关等情况,对提交给药物监管机构的药学资料的数量和程度有不同要求。
本指导原则旨在为数据的收集和报告提供更大的灵活性和避免多余的提交,简化对申办者的监管。
简化监管的二个方面如下:1、Ⅲ部分中推荐的Ⅱ期相关资料,可在Ⅱ期药物研究中生成,在Ⅱ期试验启动前无需提交。
2、Ⅳ部分中推荐的Ⅲ期相关资料,可在Ⅲ期药物研究中生成,在Ⅲ期试验启动前无需提交。
对于Ⅰ期申报,申办者可参考指导原则《药物申请Ⅰ期临床研究申报资料的格式和内容要求指导》。
本指导原则不具有法律强制性,仅阐述监管机构目前对于某个问题的看法,以及对该问题的建议。
指导原则中“应该”的字样意味着建议或推荐,而非要求。
Ⅱ. 背景A.现行法规要求根据《药品注册管理办法》,药物的临床试验必须经过国家食品药品监督管理局批准。
在每一阶段临床试验申请中,均应包含充分的药学资料,以确保其含量、质量和纯度。
提交的资料类型将取决于研究的阶段、人体研究的程度、研究周期、原料的性质与来源以及药物剂型。
目录1 品种概述 12 变更事项及变更原因 1 2.1 变更事项 1 2.2 变更内容及变更理由 12.3 变更的合理性评价和风险分析 23 变更前后辅料相关情况及其质量标准 24 制剂处方研究 3 4.1 湿法制粒处方研究 3 4.1.1 采用清膏湿法制粒 3 4.1.2 采用喷雾粉湿法制粒 4 4.2 流化床一步制粒成型研究10 4.2.1 喷雾速率考察10 4.2.2 雾化压力考察114.2.3 小试样品的制备115 生产工艺研究与验证12 5.1 湿法制粒工艺研究12 5.2 流化床一步制粒工艺研究与验证13 5.2.1 变更后XXXX工艺流程图13 5.2.2 中试工艺验证样品生产工艺14 5.2.3 3批中试工艺验证生产信息15 5.2.4 主要的生产设备16 5.2.5 各工序质量控制点16 5.2.6 中间产品的控制17 5.2.7 三批中试验证结果和评价175.3 变更前后的制备方法比较186 包装材料187 XXXX质量研究19 7.1 性状19 7.2 鉴别19 7.2.1 仪器与试药19 7.2.2 丹参薄层鉴别20 7.2.3 川芎薄层鉴别24 7.2.4 葛根素薄层鉴别28 7.3 检查32 7.3.1 水分32 7.3.2 溶化性33 7.3.3 粒度33 7.3.4 微生物限度检查337.4 含量测定35 7.4.1 仪器与试药35 7.4.2 丹参含量测定方法学验证35 7.4.3 葛根含量测定方法学验证447.4.4 XXXX(未添加蔗糖)含量测定538 变更后连续3批样品的检验报告书54 8.1 样品信息548.2 检验结果549 稳定性研究55 9.1 试验仪器55 9.2 对照品和对照药材55 9.3 试验样品56 9.4 研究内容56 9.5 研究结论57 9.6 稳定性数据58 9.6.1 加速试验58 9.6.2 长期试验621、品种概述1.1 药品名称药品名称:XXXX1.2规格:每袋装10g1.3批准文号:国药准字Z620201541.4 批准时间:2002年7月101.5执行标准国家药品标准1.6 有效期:36个月1.7 历次补充申请及最近一次再注册的情况:补充申请:2012年9月5日:企业名称由“XXXX特种药材饮片生产有限公司”变更为“XXXX 药业有限公司”,生产地址不变。
药品注册申报资料的体例与整理规范为加强药品注册纸质申报资料的规范管理,特制定本规范。
当申报资料同时进行CTD格式提交时,纸质申报资料的体例设置必须与CTD 申报格式电子文档相一致。
1.申报资料的体例要求1.1字体、字号、字体颜色、行间距离及页边距离1.1.1 字体中文:宋体英文:Times New Roman1.1.2 字号中文:不小于小4号字,表格不小于5号字;申报资料封面加粗4号;申报资料目录小4号,脚注5号字。
英文:不小于12号字。
1.1.3 字体颜色黑色1.1.4 行间距离及页边距离行间距离:单倍。
纵向页面:左边距离不小于2.5厘米、上边距离不小于2厘米、其他边距不小于1厘米。
横向页面:上边距离不小于2.5厘米、右边距离不小于2厘米、其他边距不小于1厘米。
页眉和页脚:信息在上述页边距内显示,保证文本在打印或装订中不丢失信息。
1.2纸张规格申报资料使用国际标准A4型(297mm×210mm)规格、纸张重量80g,纸张全套双面或全套单面打印,内容应完整、清楚,不得涂改。
1.3纸张性能申报资料文件材料的载体和书写材料应符合耐久性要求。
1.4加盖印章1.4.1除《药品注册申请表》、相关受理文件及检验机构出具的检验报告外,申报资料应逐个封面加盖申请人印章(多个申请人联合申报的,应加盖所有申请人印章),封面与骑缝处加盖临床研究基地有效公章,封面印章应加盖在文字处。
1.4.2 加盖的印章应符合国家有关用章规定,并具法律效力。
2.申报资料的整理要求2.1申报资料封面2.1.1申报资料袋封面2.1.1.1 档案袋封面注明:申请分类、注册分类、药品名称、本袋所属第X套第X袋每套共X袋、原件/复印件联系人、联系电话、申请单位名称。
2.1.1.2 申报资料袋封面(档案袋)应采用国家局统一格式(条码信息)的封面。
2.1.1.3 多规格的品种为同一册申报资料时,申报资料袋封面,需显示多规格的条形码的受理号(同一封面)。