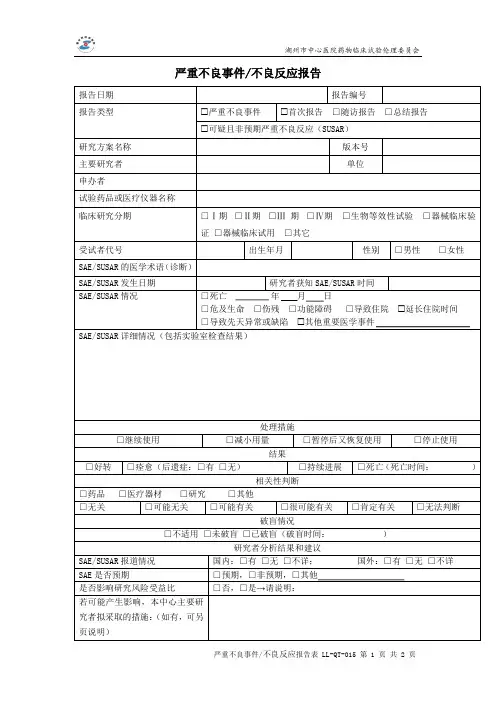
药物临床试验研究备案表-湖州中心医院
- 格式:doc
- 大小:47.50 KB
- 文档页数:2

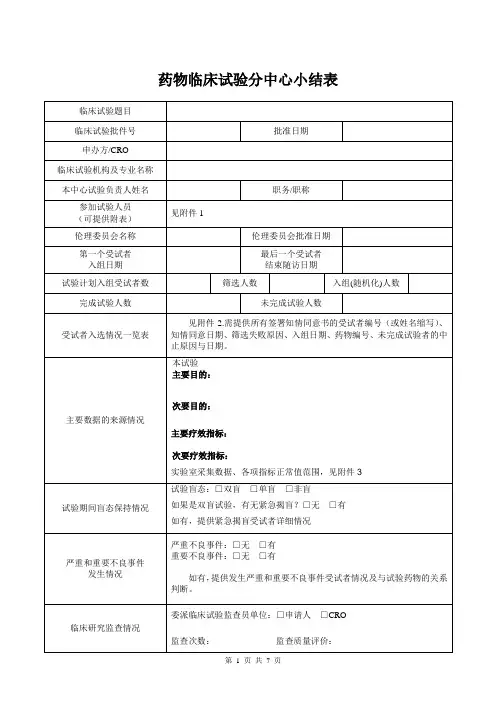
药物临床试验分中心小结表
附件1
参加试验人员信息
姓名职称所在科室研究分工
注:研究分工代码信息
1. 知情同意书获取
2. 病人入排标准确定
3. 体检/病史
4. CRF填写及更改
5. 签署原件CRF
6. 伦理联系
7. 病人联系/跟踪
8. 严重不良事件报告
9. 药物发放追踪管理
10. 试验用药指导
11. 样本管理
12. 中心实验室联系
13. IVRS/IWRS
14. 饮食和运动指导
15. 差异解决
附件2
分中心小结附表:受试者入选情况一览表
项目名称:研究中心方案号:药物名称:
第 4 页共7 页
附件3
实验室采集数据、正常值范围
第 5 页共7 页
附件4
重要不良事件
第 6 页共7 页
附件5
严重不良事件
第7 页共7 页。

药物临床试验登记填写指南(V2.0版)国家药品监督管理局药品审评中心2020年7月目录1一、简要说明 (4)2二、登记表填写指南 (6)3(一)题目和背景信息 (7)41.登记号(不可更新项、公示项) (7)52.相关登记号(可更新项、公示项) (7)63.药物名称*(不可更新项、选择公示项) (7)74.曾用名(可更新项、选择公示项) (7)85.药物类型*(不可更新项、公示项) (8)96.受理号/备案号*(不可更新项、选择公示项) (8)107.批件号(备案号)/批准日期(默示许可日期/备案日期)*(不可更11新项、不公示项) (8)128.适应症*(不可更新项、公示项) (8)139.试验专业题目*(不可更新项、公示项) (9)1410.试验通俗题目*(可更新项、公示项) (9)1511.试验方案编号*(不可更新项、公示项) (9)1612.试验方案编号重复原因(可更新项、不公示项) (9)1713.方案最新版本号*(可更新项、公示项) (9)1814.版本日期*(可更新项、公示项) (10)1915.是否联合用药*(不可更新项、公示项) (10)2016.联合用药受理号和联合用药批准证明*(不可更新项、不公示项)21 (10)22(二)申请人信息 (10)231.申请人名称(不可更新项、公示项) (10)242.联系人姓名*(可更新项、公示项) (10)253.联系人座机*(可更新项、公示项) (11)264.联系人手机号(可更新项、公示项) (11)275.联系人Email*(可更新项、公示项) (11)286.联系人邮政地址*(可更新项、公示项) (11)297.联系人邮编*(可更新项、公示项) (11)308.经费来源*(可更新项、不公示项) (11)31(三)临床试验信息 (12)321.试验目的*(不可更新项、公示项) (12)332.试验设计* (12)343.受试者信息 (15)354.试验分组 (15)365.终点指标 (17)376.数据安全监查委员会(DMC)*(可更新项、公示项) (18)387.为受试者购买试验伤害保险*(可更新项、公示项) (18)39(四)研究者信息 (19)401.主要研究者信息(可更新项、公示项) (19)412.各参加机构信息(可更新项、公示项) (20)42(五)伦理委员会信息*(不可更新项、公示项) (20)43(六)试验状态信息 (21)441.试验状态*(可更新项、公示项) (21)452.试验人数 (23)463.受试者招募及试验完成日期(不可更新项、公示项) (24)47(七)临床试验方案*(不可更新项、不公示项) (24)48(八)临床试验结果摘要(不可更新项、公示项) (25)49(九)其他附件(不可更新项、不公示项) (25)50(十)登记人及其联系方式 (25)515253一、简要说明54本指南是根据国家药品监督管理局《药品注册管理办法》(国家市场监督55管理总局令第27号)关于药物临床试验登记与信息公示的要求和药品审评中心56(以下简称“药审中心”)《药物临床试验登记与信息公示管理规范(试行)》57(以下简称“登记规范”)的要求以及对“药物临床试验登记与信息公示平台”(以58下简称“登记平台”)的总体目标要求,结合网络实现的具体设计而起草。

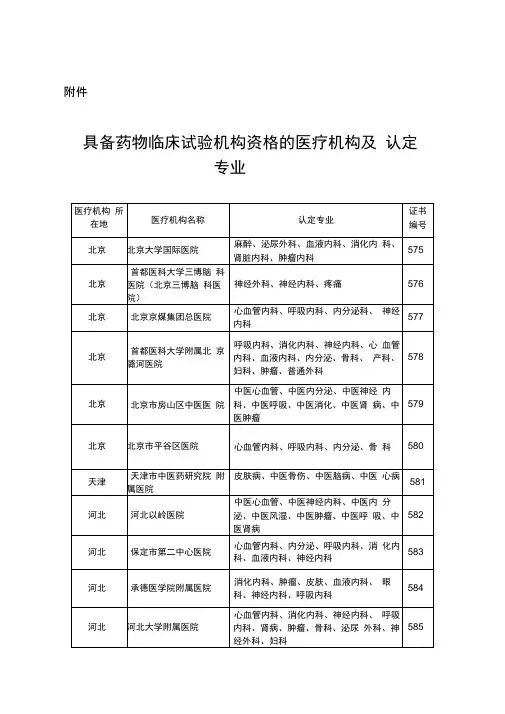
国家食品药品监督管理总局国家卫生和计划生育委员会关于药物临床试验机构开展人体生物等效性试验的公告(2017年第119号)2017年10月13日发布为落实《国务院关于改革药品医疗器械审评审批制度的意见》,更好地服务以临床价值为导向的药物创新,有效落实申请人主体责任,现就生物等效性试验有关工作公告如下:一、根据《中华人民共和国药品管理法》《药物临床试验机构资格认定办法(试行)》的有关规定,药品监督管理部门会同卫生行政部门已经认定具有药物临床试验机构资格的医疗机构619家。
经认定的药物临床试验机构均可以开展人体生物等效性试验。
二、药物临床试验机构开展人体生物等效性试验,其伦理审查和试验管理应当符合《涉及人的生物医学研究伦理审查办法》及相关指导原则中的要求、条件和程序,有效保护受试者的权益并保障其安全。
三、注册申请人开展人体生物等效性试验前,应当将拟开展的人体生物等效性试验项目在国家食品药品监督管理总局指定的化学仿制药生物等效性与临床试验备案信息平台(网址:)备案。
四、注册申请人和药物临床试验机构应当遵循《药物临床试验质量管理规范》《药物Ⅰ期临床试验管理指导原则(试行)》及相关技术要求,确保人体生物等效性试验数据真实、完整、可靠,并对全部试验数据承担法律责任。
现场检查未通过的,其数据在药品审评时将不被接受。
五、各省级药品监督管理部门负责对本行政区域内药物临床试验机构开展的人体生物等效性试验项目的监督,负责试验项目的现场检查。
对试验数据真实、完整、可靠承担监督责任。
特此公告。
附件:具有药物临床试验机构资格的医疗机构食品药品监管总局国家卫生计生委2017年9月1日2017年第119号公告附件.docx附件具有药物临床试验机构资格的医疗机构精选。


新药I 期临床试验申请技术指南(草案)国家食品药品监督管理总局(CFDA)2016年9月1目录一、前言 (1)二、背景 (2)三、与药审中心沟通交流 (3)四、IND提交所需的特定信息 (4)(一)规定的表格 (4)(二)文件目录 (5)(三)介绍性说明和总体研究计划 (5)(四)研究者手册 (6)(五)方案 (8)(六)化学、生产和控制信息 (9)(七)药理毒理信息 (14)(八)研究药物既往在人体使用的经验 (16)(九)其他重要信息 (17)(十)相关信息 (17)五、提交信息 (17)六、IND过程和审评过程 (18)(一)临床试验暂停要求 (19)(二)IND修订 (21)1七、申请人的其他责任 (22)(一)遵守法规伦理要求 (22)(二)监测正在进行的研究 (22)(三)研究药物的推销或付费 (23)(四)记录和报告 (23)(五)IND安全性报告 (23)(六)IND年度报告 (24)八、撤回、终止、暂停或者重新启动IND (25)附件 (26)附件1:药品注册临床试验申报资料信息表 (26)附件2:研究者声明表 (29)附件3: 化药Ⅰ期临床研究CMC资料表 (32)2新药I期临床试验申请技术指南一、前言国家食品药品监督管理总局(CFDA)发布本技术指南旨在帮助新药研发申请人提交足够的临床试验研究申请(Investigational new drug,IND)材料,以提高新药研发与审评效率,并同时能保证药品的安全性、有效性和质量可控性。
本指导原则阐述了在中国将研究药品(包括已进行结构确证的治疗性生物工程类产品)开始用于人体研究时,所需要提供的数据。
将申报资料的要求按照新药不同的研究阶段加以分类,在满足向CFDA提供评估拟进行研究所需要数据的同时,建立对同一类别IND申报的统一标准,增加IND申报要求的透明度、标准化,减少模糊性和不一致性,将有助于缩短新药批准进入临床试验的时间。
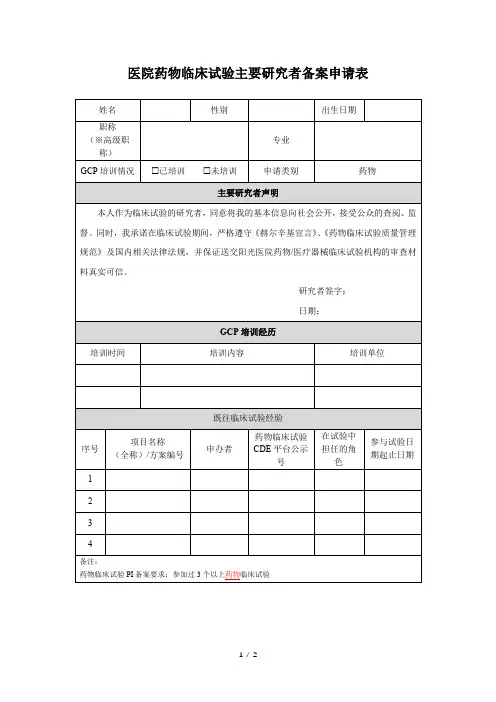

医院药物临床试验整改报告:基本药物整改报告天津市第四中心医院药物临床试验整改**国家食品药品监督管理局认证中心:2010年11月17日、18日,GCP认证检查组对我院药物临床试验机构进行了现场检查。
检查组对我院临床试验机构进行了有效、明确的指导。
针对本次检查中存在的缺陷项目,我院非常重视,安排相关岗位的人员从快、认真进行了整改,现将整改情况汇报如下:1.机构和专业需进一步完善、规范制度和标准操作规程,增强操作性;整改措施:1.1进一步细化了临床试验过程中相关的管理制度和标准操作规程,如临床试验运行管理制度、药物管理制度、文件管理制度、试验数据记录标准操作规程、严重不良事件**标准操作规程等,对临床试验的全过程都做了明确的规定,具体情况如下:1.1.1临床试验运行管理制度新修订的临床试验运行管理制度在原版基础上明确:①项目负责人在临床试验之前的职责,召开临床试验机构会议,并对临床试验相关资料进行审核。
②伦理委员会在临床试验前的职责:对临床试验相关资料进行科学审查和伦理审查。
③临床试验后项目负责人的职责:临床试验资料归档、剩余药品的退还。
以上修改处分别为临床试验运行管理制度第5、6、7、21、22条,详见附件1。
1.1.2药物管理制度新修订的药物管理制度在试验药物的交付接收环节明确专业科室如何办理试验药物的交接手续、研究者接收试验药物时的检查项目以及双盲临床试验中试验药物与对照药物的设计要求。
在试验药物的保管与记录环节中,明确试验药物如何保管以及试验药物管理人员对药物的核查。
在试验药物的分发环节明确药物管理人员如何向受试者分发药物。
增加试验药物如何退还。
以上修改处分别为药物管理制度第1.1、1.2、1.3、2.1、2.2、2.3、3.2、3.3、5.1、5.2,详见附件2。
1.1.3文件管理制度新修订文件管理制度在原版基础上增加文件的制订和修改,明确如何修订和起草临床试验相关的管理制度、设计规范、标准操作规程等文件。
西妥昔单抗治疗KRAS野生型转移性结直肠癌的效果及预后分析钟丽萍沈俊俊韩书文齐全廖海红姜亦珍潘月芬浙江省湖州市中心医院湖州师范大学附属中心医院肿瘤内科,浙江湖州313000[摘要]目的探讨西妥昔单抗治疗KRAS野生型转移性结直肠癌(mCRC)患者的治疗效果和预后因素。
方法回顾性分析2013年6月一2018年6月湖州市中心医院收治的接受西妥昔单抗治疗的83例KRAS野生型mCRC患者的临床病理学资料。
应用Cox比例风险模型对可能影响西妥昔单抗治疗无进展生存渊PFS冤时间的临床病理学因素进行单因素及多因素分析,确定独立预后因素。
结果经西妥昔单抗治疗的mCRC患者中,客观缓解率为63.9%、疾病控制率为86.7%。
全组中位随访15.5个月,中位PFS为8.7个月,6、12、18个月PFS率分别为71.1%、32.5%、13.3%。
多因素分析显示肿瘤原发部位、基线糖类抗原(CA)199水平、是否发生皮疹及严重程度及早期肿瘤退缩是患者预后的独立影响因素(P<0.05)o结论基线CA199水平、肿瘤位置、皮疹、早期肿瘤退缩可能是西妥昔单抗治疗KRAS野生型mCRC患者PFS的独立危险因素,有助于预测西妥昔单抗治疗的效果和预后。
[关键词]西妥昔单抗;转移性结直肠癌;效果;预后冲图分类号]R735.3[文献标识码]A[文章编号]1673-7210(2020)12(b)-0104-04Efficacy and prognostic analysis of Cetuximab in the treatment of KRAS wild-type metastatic colorectal cancerZHONG Liping SHEN Junjun HAN Shutven QI Quan LIAO Haihong JIANG Yizhen PAN Yuefen DeparLmenL of Medical Oncology,Huzhou CenLral HospiLal AffiliaLed HospiLal of Huzhou Normarl UniversiLy,Zhejiang Province,Huzhou313000,China[Abstract]Objective To explore Lhe efficacy and prognosLic facLors of CeLuximab in Lhe LreaLmenL of KRAS wild-Lype meLasLaLic colorecLal cancer(mCRC)paLienLs.Methods A reLrospecLive analysis of Lhe clinicopaLhological daLa of83 cases of KRAS wild-Lype mCRC paLienLs LreaLed wiLh CeLuximab in Huzhou CenLral HospiLal from June2013Lo June 2018.The COX proporLional hazard model was used Lo conducL Lhe univariaLe and mulLivariaLe analyses of Lhe clinico-paLhological facLors LhaL may influence Lhe LreaLmenL of progression free survivals(PFS)Lime wiLh ceLuximab Lo deLer-mine Lhe prognosLic facLors.Results Among paLienLs wiLh mCRC LreaLed wiLh ceLuximab,Lhe objecLive response raLe was 63.9%,and Lhe disease conLrol raLe was86.7%.The median follow-up for Lhe whole group was15.5monLhs,Lhe median PFS was8.7monLhs,and Lhe PFS raLes aL6,12,and18monLhs were71.1%,32.5%and13.3%,respecLively.MulLi-variaLe analysis showed LhaL Lhe primary Lumor siLe,baseline carbohydraLe anLigen(CA)199level,rash and severiLy and early Lumor regression were independenL prognosLic facLors(P<0.05).Conclusion Baseline CA199level,Lumor locaLion,skin rash,and early Lumor regression may be independenL risk facLors for PFS in paLienLs wiLh KRAS wild-Lype mCRC LreaLed wiLh ceLuximab,and help predicL Lhe effecL and prognosis of ceLuximab LreaLmenL.[Key words]CeLuximab;MeLasLaLic colorecLal cancer;Efficacy;Prognosis结直肠癌是全球最常见的恶性肿瘤之一,也是导致癌症死亡的主要原因[1]o转移性结直肠癌(metastatic colorectal cancer,mCRC)的预后不良,5年生存率仅为13.3%[2]。