药品不良反应报告和监测检查资料清单
- 格式:docx
- 大小:14.21 KB
- 文档页数:4

药品不良反应报告和监测检查指南(试行)药品不良反应报告和监测是药品上市后监管的重要内容,是药品生产企业对其生产的药品进行全生命周期管理的主要内容和重要责任,是药品安全评价的重要依据。
为推进药品生产企业开展不良反应报告和监测工作,指导食品药品监督管理部门开展对企业药品不良反应报告和监测工作的检查,制定本检查指南。
本指南适用于食品药品监督管理部门开展对药品生产企业不良反应报告和监测工作的检查。
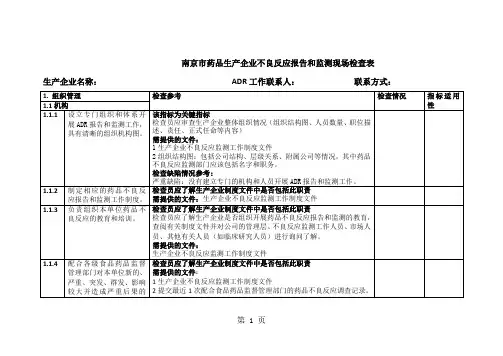
1.检查目的1.1通过对药品生产企业执行《药品不良反应报告和监测管理办法》情况进行检查,促进企业规范开展药品不良反应报告和监测工作,推进相关工作全面开展。
1.2确定药品生产企业具备符合法律法规所要求的机构、人员、制度、体系和设施。
1.3检查药品生产企业是否严格履行报告和监测责任,是否存在可能对公众健康造成威胁的因素和风险。
1.4必要时,检查结果可作为监管措施的依据。
2.检查类型2.1常规检查:指按计划开展的例行检查,通常为系统性检查,可选取一个或多个特定药品作为实例检查企业药品不良反应报告和监测体系运转情况。
常规检查应坚持问题导向,如新药监测期内、首次进口5年内的药品为重点检查内容。
常规检查范围包括组织机构、人员管理、质量管理体系、个例药品不良反应、药品群体不良事件、境外发生的严重药品不良反应、定期安全性更新报告、药品重点监测、评价及控制9个方面,检查内容参见检查要点(见附件1)。
疫苗生产企业的个例报告、群体不良事件检查内容,参考《全国疑似预防接种异常反应监测方案》(卫办疾控发〔2010〕94号)有关要求。
2.2有因检查:指因一个或多个特殊问题,对生产企业开展的针对性检查,以确认生产企业是否存在违反相关法规的行为。
启动有因检查原因如下:2.2.1药品安全性出现问题:未能及时发现或沟通相关风险,产品撤市或暂停生产、销售时未提前告知等。
2.2.2报告责任未履行:迟报、瞒报、报告质量差、报告信息不准确等。
2.2.3未完成监管机构的要求:未按要求开展药品重点监测;未按要求实施相关产品的风险管理计划;未按要求提供资料,或提供的数据信息不符合要求。

药品不良反应报告和监测检查指南(试行)药品不良反应报告和监测是药品上市后监管的重要内容,是药品生产企业对其生产的药品进行全生命周期管理的主要内容和重要责任,是药品安全评价的重要依据。
为推进药品生产企业开展不良反应报告和监测工作,指导食品药品监督管理部门开展对企业药品不良反应报告和监测工作的检查,制定本检查指南。
本指南适用于食品药品监督管理部门开展对药品生产企业不良反应报告和监测工作的检查。
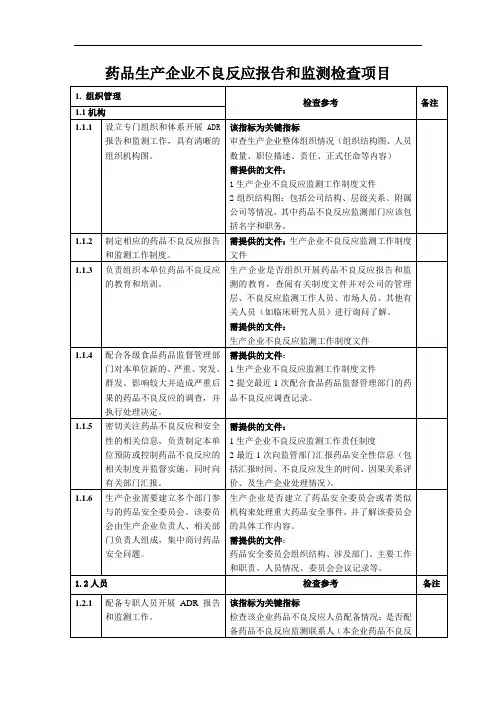
1.检查目的1.1通过对药品生产企业执行《药品不良反应报告和监测管理办法》情况进行检查,促进企业规范开展药品不良反应报告和监测工作,推进相关工作全面开展。
1.2确定药品生产企业具备符合法律法规所要求的机构、人员、制度、体系和设施。
1.3检查药品生产企业是否严格履行报告和监测责任,是否存在可能对公众健康造成威胁的因素和风险。
1.4必要时,检查结果可作为监管措施的依据。
2.检查类型2.1常规检查:指按计划开展的例行检查,通常为系统性检查,可选取一个或多个特定药品作为实例检查企业药品不良反应报告和监测体系运转情况。
常规检查应坚持问题导向,如新药监测期内、首次进口5年内的药品为重点检查内容。
常规检查范围包括组织机构、人员管理、质量管理体系、个例药品不良反应、药品群体不良事件、境外发生的严重药品不良反应、定期安全性更新报告、药品重点监测、评价及控制9个方面,检查内容参见检查要点(见附件1)。
疫苗生产企业的个例报告、群体不良事件检查内容,参考《全国疑似预防接种异常反应监测方案》(卫办疾控发〔2010〕94号)有关要求。
2.2有因检查:指因一个或多个特殊问题,对生产企业开展的针对性检查,以确认生产企业是否存在违反相关法规的行为。
启动有因检查原因如下:2.2.1药品安全性出现问题:未能及时发现或沟通相关风险,产品撤市或暂停生产、销售时未提前告知等。
2.2.2报告责任未履行:迟报、瞒报、报告质量差、报告信息不准确等。
2.2.3未完成监管机构的要求:未按要求开展药品重点监测;未按要求实施相关产品的风险管理计划;未按要求提供资料,或提供的数据信息不符合要求。
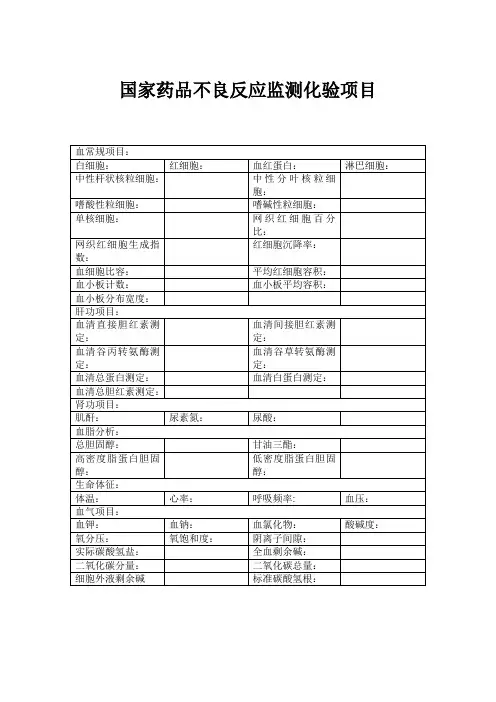
国家药品不良反应监测化验项目
血常规项目:
白细胞:红细胞:血红蛋白:淋巴细胞:中性杆状核粒细胞:中性分叶核粒细
胞:
嗜酸性粒细胞:嗜碱性粒细胞:
单核细胞:网织红细胞百分
比:
网织红细胞生成指
数:
红细胞沉降率:
血细胞比容:平均红细胞容积:
血小板计数:血小板平均容积:
血小板分布宽度:
肝功项目:
血清直接胆红素测定:血清间接胆红素测定:
血清谷丙转氨酶测定:血清谷草转氨酶测定:
血清总蛋白测定:血清白蛋白测定:血清总胆红素测定:
肾功项目:
肌酐:尿素氮:尿酸:
血脂分析:
总胆固醇:甘油三酯:
高密度脂蛋白胆固醇:低密度脂蛋白胆固醇:
生命体征:
体温:心率:呼吸频率: 血压:血气项目:
血钾:血钠:血氯化物:酸碱度:氧分压:氧饱和度:阴离子间隙:
实际碳酸氢盐:全血剩余碱:
二氧化碳分量:二氧化碳总量:
细胞外液剩余碱标准碳酸氢根:。

药物不良反应报告表1. 背景药物不良反应报告表是用于记录患者或医疗专业人员对药物使用过程中出现的不良反应的表格。
通过收集和整理这些不良反应数据,可以帮助监测药物的安全性和有效性,并及时采取相应的措施保护患者的健康安全。
2. 表格字段药物不良反应报告表应包含以下字段:- 患者信息:包括患者姓名、性别、年龄和联系方式等个人基本信息。
- 药物信息:包括药物名称、剂量、使用频率和使用时间等药物使用相关信息。
- 不良反应描述:详细描述患者所出现的不良反应,包括症状、出现时间和持续时间等详细信息。
- 严重程度评估:对不良反应的严重程度进行评估,可以采用轻、中、重等级别进行分类。
- 处理措施:记录医疗专业人员采取的处理措施,如停药、调整剂量或联系医生等。
3. 使用说明填写药物不良反应报告表时,应注意以下事项:- 填写者应提供真实准确的信息,确保数据的可靠性。
- 在描述不良反应时,尽量使用客观的、准确的语言。
避免主观评价和夸大描述。
- 对于不良反应的严重程度评估,应根据医疗专业人员的经验和相关标准进行判断。
- 在记录处理措施时,要详细说明具体的操作步骤,以便日后参考和追踪。
4. 数据分析和应用收集到的不良反应报告表可以进行数据分析,得出以下信息:- 药物的常见不良反应类型和发生率。
- 不同药物在不同人群中的安全性和耐受性情况。
- 不同药物的严重不良事件是否存在差异。
- 不同处理措施对不良反应的有效性比较。
通过对这些信息的分析和应用,可以为药物安全监测和临床决策提供科学依据,进一步提高医疗质量和患者的用药安全性。
5. 结论药物不良反应报告表是记录和收集药物不良反应信息的重要工具,能够帮助监测药物的安全性和有效性,并为临床决策提供依据。
正确使用和填写该表,能够提高药物安全性监测的效果,保障患者健康安全。



药物警戒检查指导原则为指导药品监督管理部门开展药物警戒检查工作,督促药品上市许可持有人(以下简称持有人)落实药物警戒主体责任,根据《药品检查管理办法(试行)》等有关规定,制定本指导原则。
本指导原则适用于省级及以上药品监督管理部门对持有人自行开展及其委托开展的药物警戒活动进行的检查工作;对获准开展药物临床试验的药品注册申请人开展药物警戒检查的,应结合药物安全性特性和临床试验安全信息报告及风险评估,在临床试验期间或上市许可前启动药物警戒检查,具体实施可参照本指导原则。
有关检查工作的组织实施,以及检查机构和人员、检查程序、常规检查、有因检查、检查与稽查的衔接、跨区域检查协作、检查结果的处理等相关工作,按照《国家药监局关于印发〈药品检查管理办法(试行)〉的通知》(国药监药管〔2021〕31号)等有关要求执行。
一、常规检查重点考虑因素(一)药品特征1.药品的安全性特性。
2.药品不良反应监测数据及药品不良反应聚集性事件发生情况。
3.销售量大或替代药品有限的药品。
4.批准上市时有附加安全性条件的药品。
—1—5.创新药、改良型新药,以及针对儿童、孕产妇等特殊群体使用的药品。
6.社会关注度较高的药品。
(二)持有人特征7.持有品种较多、销售量大的持有人。
8.未接受过药物警戒检查的持有人。
9.首次在中国境内获得药品注册证书的持有人。
10.企业发生并购、组织结构变更等导致药物警戒体系发生重大变化或对药物警戒组织结构有重大影响的持有人。
11.委托生产的持有人。
12.委托开展药物警戒活动的持有人。
(三)其他情况13.既往药物警戒检查或其他检查情况。
14.药品监督管理部门认为需要开展检查的其他情况。
二、有因检查重点考虑因素(一)对疑似药品不良反应信息迟报、瞒报、漏报,报告质量差的。
(二)药品不良反应监测提示可能存在安全风险的。
(三)未能及时发现、评估、控制或沟通相关风险的。
(四)采取暂停生产、销售、使用和产品召回,未按规定报告药品监督管理部门的。

药品不良反应登记清单
1. 背景信息
药品不良反应发生后,及时记录和登记是保护患者安全的重要措施。
本登记清单旨在提供一个便捷的工具,用于记录和追踪药品的不良反应情况。
2. 登记要求
在药品不良反应发生时,下列信息应被登记:
- 患者个人信息:姓名、性别、年龄
- 药品信息:药品名称、批号、剂量、给药途径、用药频次、用药期间
- 不良反应描述:详细描述不良反应的症状、严重程度等
- 其他相关信息:治疗措施、反应持续时间、是否需要住院治疗等
登记表格应具备以下要点:
- 表格应简洁明了,易于填写和阅读
- 每一次不良反应应在单独的行中登记
- 表格应采用规范的日期格式和统一的单位
- 表格应具备编号或唯一标识,方便排序和追踪记录
3. 数据分析和追踪
登记表格收集的不良反应数据可用于进一步分析和追踪。
以下是一些可能的数据分析方向:
- 不良反应的发生率和严重程度趋势分析
- 不同药品的不良反应比较
- 不良反应与患者特征的相关性分析
数据分析和追踪结果应及时报告给相关部门和决策者,以便采取必要的措施,确保药物的安全使用。
4. 数据保密和隐私
在进行数据收集和使用过程中,要确保患者个人信息的保密和隐私。
以下是一些保密和隐私措施:
- 仅限授权人员可访问登记表格和相关数据
- 培训工作人员,提醒他们妥善处理敏感信息
- 遵守相关法律和法规,确保数据保护和隐私权
5. 结论
药品不良反应登记清单是记录和追踪药品不良反应的重要工具。
通过准确记录和及时追踪,可以更好地保护患者的安全和健康。
同时,要确保数据的保密和隐私,以保护患者的个人信息不被泄露。
药品不良反应报告和监测检查资料清单
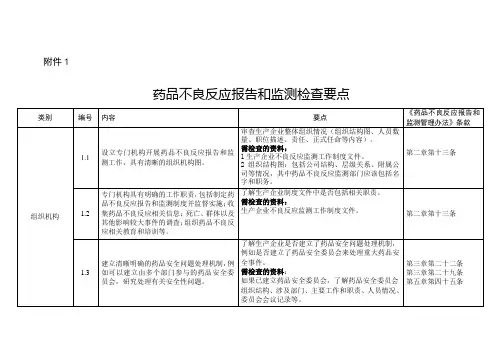
1.基本信息
1.1生产企业联系人联系方式(电话、电子邮箱、传真、通信地址、邮编等)。
1.2药品注册信息:生产企业药品注册情况列表,包括注册时间、上市时间、是否新药监测期品种、说明书变更、撤市、暂停、召回等情况等。
1.3 药品不良反应监测工作概况:描述药品不良反应监测工作如何有效开展,如工作流程、各部门之间协调管理(包括与全球或总部相关部门的协调)、人员安排等。
2.组织机构
2.1组织结构图:包括附属公司及机构情况,其中药品不良反应监测部门应该包括工作人员姓名和职务。
2.2 药品不良反应监测部门职责。
2.3 药品安全问题处理机制
3.人员管理
3.1 药品不良反应监测专职人员职责描述、专业背景、培训记录等。
3.2 药品不良反应报告相关部门人员培训记录。
4.质量管理体系
4.1管理制度:药品不良反应报告和监测、人员培训、资料管理等制度文件。
4.2程序文件须详细描述下述药品不良反应监测工作流程。
4.2.1个例药品不良反应报告处理。
4.2.2药品群体不良事件处理。
4.2.3境外发生的严重药品不良反应处理。
4.2.4定期安全性更新报告。
4.2.5药品重点监测。
4.2.6药品安全性信号检测。
4.2.7说明书更新程序。
4.2.8对于药品监管机构提出问题的回复程序。
4.2.9处理医学咨询和投诉程序。
4.2.10 文献检索程序。
4.2.11 评价与控制程序。
4.2.12数据处理程序:包括数据收集、整理,如有电子收集系统,应描述系统版本、系统的支持和维护等。
4.2.13资料存档:文件(包括电子文档)的归档和存储程序。
4.3质量管理体系审核:近一年内部审核和/或外部审核的总结报告、问题清单、整改情况等。
5.个例药品不良反应报告
个例药品不良反应报告汇总,包括年度药品不良反应报告总数、严重及死亡报告情况。
6.药品群体不良事件报告
6.1药品群体不良事件列表,包括药品名称、报告编码、报告日期、累及人数、不良反应表现、不良反应结果、关联性评价等信息。
6.2药品群体不良事件案例1起,内容包括
《药品群体不良事件报告表》、《药品不良反应/事件报告表》、调查报告等。
7.境外发生的严重药品不良反应报告
境外发生的严重药品不良反应汇总,包括药品名称、年度严重报告数量、及时报告比例。
8.定期安全性更新报告
定期安全性更新报告情况列表,包括药品名称、报告周期、最近一次报告情况等。
9.药品重点监测
9.1根据《药品不良反应报告和监测管理办法》,说明应开展药品重点监测的品种名称、实施情况等。
9.2 药品重点监测案例1个,内容包括实施方案、中期报告、总结
报告等。
10.评价及控制
10.1 信号检测、监测数据定期分析(含国家药品不良反应监测机构反馈数据)工作开展情况,并提供相关案例1个。
10.2药品上市后研究开展情况:包括药品名称、研究时间、研究目的、研究方法、研究结果等;提供相关案例1个。
10.3发现的药品主要安全性问题以及采取的风险控制措施(包括制定风险管理计划)情况,并提供相关案例1个。
11.其他
11.1 生产企业药品不良反应报告和监测工作自查情况,并描述存在的问题情况。
11.2生产企业需要说明的其他问题(如,与药品不良反应监测工作有关的委托项目情况等)。
备注:
1. 检查资料的准备请参考“药品不良反应报告和监测检查项目”。
2.有关资料或列表汇总期限、涉及的药品、提交资料数量等具体情况,视检查需要确定。
3.上述资料均为中文,并请在每项资料左上角标注“生产企业名称+资料X”(X为资料编号)。