基于16SrRNA_DNA分析的土壤微生物生态学效应_黄进勇
- 格式:pdf
- 大小:113.01 KB
- 文档页数:4


基于拟杆菌16S rRNA基因进行微生物溯源的研究进展
梁红霞;余志晟;刘如铟;张洪勋;吴钢
【期刊名称】《中国环境科学》
【年(卷),期】2018(38)11
【摘要】微生物溯源方法可利用粪便中的微生物区分来自人或动物的粪便污染.其中,拟杆菌以其丰度高、不能体外繁殖和宿主特异性强等优势被广泛应用于微生物溯源研究中.本文以拟杆菌16S rRNA基因为标记物,总结了拟杆菌及其标记物在环境中的衰减、拟杆菌引物的敏感性和特异性以及分子生物学技术在微生物溯源中的运用,可为粪便污染源解析提供一定的科学参考依据.
【总页数】10页(P4236-4245)
【作者】梁红霞;余志晟;刘如铟;张洪勋;吴钢
【作者单位】中国科学院大学资源与环境学院,北京100049;中国科学院大学资源与环境学院,北京100049;中国科学院大学资源与环境学院,北京100049;中国科学院大学资源与环境学院,北京100049;中国科学院生态环境研究中心,北京100085【正文语种】中文
【中图分类】X172
【相关文献】
1.16S rRNA基因序列在分枝杆菌分类鉴定的研究进展 [J], 刘佳文;吕红艳;吴雪琼
2.基于16S/18S rRNA基因文库技术研究酿醋大曲固态发酵过程中微生物群落结构演变 [J], 刘雄;甘兴;罗立新
3.基于16S rRNA基因分析双斑东方鲀肠道
微生物多样性 [J], 雷阳;王松刚;陈钰;姜尧;郑毅
4.基于拟杆菌特异性16S rRNA基因的塘坝型饮用水污染溯源研究 [J], 张曦;朱昌雄;冯广达;朱红惠;郭萍
5.基于16S rRNA基因的枯草芽孢杆菌PCR快速检测方法的建立 [J], 李天芝;于新友;沈志强
因版权原因,仅展示原文概要,查看原文内容请购买。

红树林土壤细菌和古菌的16S红树林土壤中细菌和古菌的16S基因研究:揭示物种多样性与环境适应性红树林是一种独特的生态系统,富含生物多样性。
其中,土壤微生物在其生态功能的发挥中扮演着重要角色。
作为生态系统的重要组成部分,土壤细菌和古菌的16S基因研究对于了解红树林的生态特性具有重要意义。
本文旨在探讨红树林土壤细菌和古菌的16S基因,进一步理解其物种多样性与环境适应性。
在过去的研究中,已有多位学者对红树林土壤细菌和古菌的16S基因进行了深入研究。
他们通过基因测序技术,揭示了红树林土壤中细菌和古菌的物种多样性以及不同环境下的序列差异。
这些研究还对红树林土壤微生物的进化关系进行了探讨,并对其功能进行了初步注释。
本研究采用Illumina MiSeq平台进行测序,对红树林土壤细菌和古菌的16S基因进行测序分析。
我们收集了不同红树林土壤样本,提取其总DNA。
然后,通过PCR扩增16S基因片段,并进行纯化和建库。
采用MiSeq平台进行测序,得到原始序列数据。
通过分析原始序列数据,我们得到了红树林土壤细菌和古菌的16S基因序列。
进一步的数据处理和可视化分析显示,红树林土壤中存在着丰富的细菌和古菌物种多样性,且不同红树林间的物种组成存在一定的差异。
我们还发现了一些具有特殊适应性的细菌和古菌,例如能够在高盐度环境下生存的物种。
这些发现为我们深入了解红树林土壤微生物的生态功能提供了基础。
我们的研究发现,红树林土壤细菌和古菌的16S基因序列在不同红树林间存在差异,这可能与它们对环境的适应性有关。
例如,某些特殊环境下生存的物种可能在其生境中具有更高的竞争力和生存能力。
我们还发现了一些与环境特性密切相关的功能注释,进一步证实了这些微生物在红树林生态系统中的重要功能。
这与先前的研究结果相一致,进一步证实了我们的结论。
通过对红树林土壤细菌和古菌的16S基因研究,我们发现其物种多样性与环境适应性之间存在密切关系。
这些发现不仅有助于我们更好地理解红树林生态系统的运行机制,也为今后研究提供了重要的参考依据。

基于16SrRNA测序技术的微生物组多样性分析研究第一章入门概述在微生物学领域,了解微生物组的多样性和组成的重要性越来越受到关注。
通过对微生物组中微生物物种的鉴定和数量的测定,能够帮助我们了解微生物在不同环境中的生态功能及其对宿主的影响。
而基于16SrRNA测序技术的微生物组研究,可以直接从微生物的遗传信息入手,不但速度快、准确性高,而且可以在不同层次上研究微生物组的多样性。
本文主要介绍基于16SrRNA测序技术的微生物组多样性分析研究的基本原理、方法以及相应的应用。
第二章基本原理16S rRNA是所有细菌和古菌都具备的一种RNA,位于小亚基的30S核糖体亚基上。
其序列具有高度的起始和终止保守性,而在两端之间则存在一定的变异性。
早期16S rRNA序列的研究表明,它的高度保守性和存在变异性的特点可以用来鉴定不同的细菌和古菌,从而对微生物的分类、系统发育和进化关系进行研究。
近年来,随着高通量测序技术的不断发展,人们开始采用基于16SrRNA的高通量测序技术来研究微生物组的多样性和组成。
基于16SrRNA测序技术的微生物组研究,主要基于对微生物16SrRNA基因序列的测序和分析。
这种技术可以通过对不同体系样本中的微生物16SrRNA基因进行PCR扩增,获得大量的16SrRNA序列信息,从而对微生物组的多样性和组成进行深入研究。
第三章方法流程基于16SrRNA测序技术的微生物组分析方法主要包括样品采集、总DNA提取、16SrRNA基因的PCR扩增、高通量测序、序列质量控制、OTU聚类、分类学注释和多样性分析等若干步骤。
1. 样品采集微生物组研究的第一步是采集样品。
不同的研究目的和需要不同的样品类型,快速、高效、准确的样品采集对于后续的研究至关重要。
对于土样等环境样品,我们应该考虑样品中微生物细胞在不同深度的分布情况,采集不同深度的样品,尽可能地覆盖样品中的全部微生物种群。
对于丰富样品,如粪便、口腔、皮肤表面等,我们需要考虑样品中微生物群落在患者或者宿主的年龄、性别、健康状态等方面差异,采集不同个体的样品进行相应比较。

土壤微生物群落及其功能的高通量测序技术研究土壤微生物群落是指土壤中的各种微生物(包括细菌、真菌、古菌和叶绿体、线粒体等小型生物)的总称。
它们以其丰富的分类、种群丰度和多样性,被认为是维持土壤生态系统功能和养分循环的关键因素。
因此,研究土壤微生物群落及其功能对于理解土壤生态系统功能和土壤质量的影响具有重要意义。
随着高通量测序技术的发展,特别是基于16SrRNA等功能基因的测序技术,研究土壤微生物群落及其功能的研究进展迅速。
高通量测序技术通过从土壤样品中提取DNA或RNA,利用PCR扩增目标基因片段,并通过高通量测序仪器获取海量的序列数据。
这些数据可以揭示出土壤微生物的多样性、丰度、功能以及微生物间的相互作用等信息。
研究土壤微生物群落及其功能的高通量测序技术主要包括以下几个方面:1. 多样性分析:通过对测序数据进行聚类、物种多样性指数计算和Beta多样性分析等统计方法,可以了解土壤微生物群落的物种组成、多样性和相对丰度等信息。
2.功能注释:通过比对测序数据与数据库中的功能基因序列进行比对,可以对土壤微生物的功能进行注释,如其参与的代谢途径、功能基因的存在与否等。
3.生态功能研究:通过分析测序数据中的功能基因组成和它们的相互作用网络等信息,可以预测土壤微生物群落对养分循环、有机物降解、抗性基因传播等功能的影响。
4. 基因表达研究:通过转录组测序(RNA-seq)等技术,可以研究土壤微生物群落在不同环境条件下的基因表达变化,以及参与重要生态功能的关键基因的表达情况。
5.网络分析和预测模型:通过将多个生态学和统计学方法相结合,可以构建微生物间相互作用网络,预测土壤微生物群落的养分循环、有害物分解等功能,并为土壤生态系统的管理和保护提供决策支持。
总之,高通量测序技术在研究土壤微生物群落及其功能方面具有重要的应用潜力。
通过揭示土壤微生物群落的多样性、丰度、功能以及微生物间的相互作用,可以深入理解土壤生态系统的功能和稳定性,并为保护、修复和提高土壤质量提供科学依据。

16srrna基因技术在油藏微生物生态研究中的应用
16S rRNA基因技术是一种用于微生物分类和地球环境微生物
群落结构研究的重要方法。
在油藏微生物生态研究中,16S rRNA基因技术被广泛应用于发现、鉴定和研究油藏中的微生
物群落。
首先,通过对油藏中微生物群落的16S rRNA基因进行测序和
分析,可以获得不同微生物群落的组成和多样性信息。
这有助于了解油藏中微生物的种类、数量和相对丰度等关键生态信息,从而揭示微生物群落的结构和功能。
其次,16S rRNA基因技术可以用于鉴定油藏中的微生物。
通
过比对16S rRNA基因序列数据库,可以确定微生物的分类和
系统发育位置,从而识别油藏中存在的不同微生物种类。
此外,16S rRNA基因技术还可以帮助研究油藏中微生物的代
谢功能和生态作用。
通过分析16S rRNA基因的功能区域,可
以推断微生物的功能特性,如产酸、产气、产聚合物降解酶等,从而揭示微生物对油藏生态系统中有机物转化、油藏酸化和生物降解等方面的影响。
综上所述,16S rRNA基因技术在油藏微生物生态研究中起着
重要作用。
它可以提供关于微生物群落结构、种类、数量和功能等方面的重要信息,有助于深入理解油藏微生物的生态特性和作用机制,为油藏开发和油藏环境管理提供科学依据。

16S rRNA论文疾病诊断论文环境保护论文:16S rRNA基因应用研究进展【摘要】16s rrna分析的实验技术为微生物生态学的研究提供了新的研究方法,因此越来越多的学者利用16s rrna 进行各种实验的研究并取得了骄人的成绩。
本文对16s rrna 基因的获得做一简单介绍,重点介绍16s rrna基因的应用。
16s rrna在细菌菌种鉴定、环境保护、疾病诊断等方面都取得了很好的研究进展,不久的将来16s rrna基因将会为人类做出更大的贡献。
【关键词】16s rrna;菌种鉴定;疾病诊断;环境保护前言随着遗传学、分子生物学、细胞生物学等学科的迅速发展,越来越多的新方法和新技术被应用于分子水平研究领域。
在这样的背景下,16s rrna基因的研究无论在广度和深度上都取得了显著的成就,16s rrna基因技术的应用在微生物多样性、微生物种群分析、重要基因的发现、以及遗传物质在微生物之间或微生物与非生物环境之间相互关系等方面均做出了巨大的贡献,并开辟了微生物生态学研究新的领域。
关于16s rrna基因方面的综合性论述尚未见报道,本文将为该领域的进一步研究提供必要的综合信息。
1 16s rrna基因的简介:细菌rrna按沉降系数分为3种,分别为5s、16s和23s rrna。
其中位于原核细胞核糖体小亚基上的16s rrna长约1540bp,结构和碱基排列复杂度适中,较易于进行序列测定和分析比较。
16s rdna是细菌染色体上编码16s rrna相对应的dna序列,存在于所有细菌染色体基因中,它的内部结构由保守区及可变区两部分组成,因此可用pcr扩增其相应的rdna片断,来快速、灵敏地检测样品中是否存在某些细菌或致病菌,或进行细菌多样性分析。
2 16s rrna基因在细菌菌种鉴定中的应用:1977年c.woese通过对各种生物的rrna进行分析,认为16s rrna 基因及其类似的rrna基因序列作为生物系统发育指标最为合适,提出了可将自然界的生命分为细菌、古菌和真核生物三域(domain),揭示了各生物之间的系统发育关系,使微生物学进入到成熟时期,因此许多研究采用测16s rrna基因部分序列的方法进行多样性分析。

土壤细菌16SrRNA基因变异型及其与植被的相关研究
杨官品;朱艳红;陈亮;薛小乔
【期刊名称】《应用生态学报》
【年(卷),期】2001(12)5
【摘要】绕过细菌的分离培养 ,直接提取土壤DNA ,扩增、克隆土壤细菌群体的16S核糖体RNA基因 (16SrD NA) .根据该基因各种变异类型的限制性片段长度多型性 ,分析土壤细菌分子遗传多样性及其与植被的相互关系 .植被的改变影响土壤养分 ,进而改变土壤细菌群落结构 .土壤细菌遗传多样性和分化能反映植被的变化 .【总页数】4页(P757-760)
【关键词】细菌;16S核糖体RNA基因;限制性片段长度多型性;植被;变异类型;土壤养分
【作者】杨官品;朱艳红;陈亮;薛小乔
【作者单位】湖北大学生命科学学院
【正文语种】中文
【中图分类】Q933;S158.381
【相关文献】
1.16SrRNA及相关技术用于临床细菌鉴定的研究进展 [J], 叶乃芳(综述);王中新(审校)
2.高寒草甸群落地表植被特征与土壤理化性状、土壤微生物之间的相关性研究 [J], 王长庭;龙瑞军;王根绪;刘伟;王启兰;张莉;吴鹏飞
3.CPPS患者前列腺液细菌16SrRNA基因及白细胞数与临床疗效的相关性研究[J], 陈修德;金讯波;夏庆华;赵勇;蒋绍博;郑宝钟
4.基于16SrRNA的肝郁血虚型失眠患者口腔微生态与中医舌象的相关性研究 [J], 刘梦;王曦廷;李峰;谭丽博;李杰;关静
5.土壤细菌遗传多样性及其与植被类型相关性研究 [J], 杨官品;男兰;贾海波;朱艳红;刘英杰;张凯
因版权原因,仅展示原文概要,查看原文内容请购买。
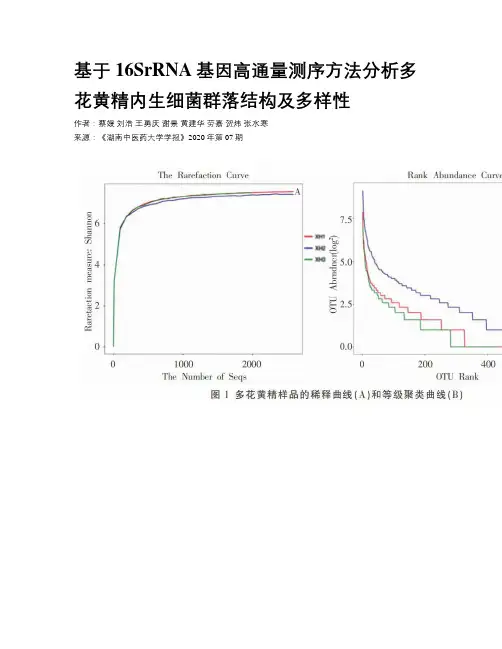
基于16SrRNA基因高通量测序方法分析多花黄精内生细菌群落结构及多样性作者:蔡媛刘浩王勇庆谢景黄建华劳嘉贺炜张水寒来源:《湖南中医药大学学报》2020年第07期〔摘要〕目的采用高通量測序技术分析多花黄精内生细菌的多样性及菌群结构。
方法通过蒽酮-硫酸法测定多糖含量,采用通用引物对细菌16S rRNA高可变区(V3-V4区)进行PCR扩增,运用Illumina Miseq PE250高通量测序技术对扩增子进行测序,并用QIIME等软件对测序序列进行生物信息学分析。
通过Spearman相关性分析菌群与多糖含量的相关性。
结果多花黄精内生菌测序共获得90 628条有效序列和5 287个OTU,稀释曲线和Coverage指数反映测序结果比较全面地覆盖了多花黄精内生细菌群落。
Alpha多样性分析表明,其内生细菌多样性程度高。
在门水平,其优势菌门为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、芽单胞菌门(Gemmatimonadetes)、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)。
属水平,核心菌群由鞘氨醇单胞菌属(Sphingomonas)、芽单胞菌属(Gemmatimonas)、链霉菌属(Streptomyces)组成。
PICRUSt基因预测表明,多花黄精内生细菌以代谢功能为主,主要包括能量代谢、碳水化合物代谢、氨基酸代谢等。
通过Spearman 相关性分析,在属水平共得到19个菌群与多糖含量相关,其中3个属呈正相关,16个属菌群呈负相关。
结论多花黄精内生细菌的多样性程度高,存在多种不同种类的细菌,且菌群与多糖含量相关。
该研究解析了多花黄精内生细菌的多样性、丰度及主要菌属,对深入研究多花黄精内环境有一定指导意义。
〔关键词〕多花黄精;16S rRNA;内生细菌;多样性;群落结构〔中图分类号〕R284.1 〔文献标志码〕A 〔文章编号〕doi:10.3969/j.issn.1674-070X.2020.07.013〔Abstract〕 Objective To analyze the bacterial diversity and their community structure from Polygonatum cyrtonema using high-throughput sequencing technology. Methods The polysaccharide content was determined by anthrone-sulfuric acid method. PCR amplification of 16S rRNA high variable region (V3/V4 region) of bacteria was performed by using universal primers. The sequencing of amplicons was conducted by using Illumina Miseq PE250 high-throughput sequencing technology, and bioinformatics analysis of sequencing sequences was performed using software such as QIIME. The correlation between endophytic bacteria and polysaccharide was analyzed by spearman correlation. Results A total of 90 628 effective sequences and 5 287 OTUs were obtained from the sequencing of endophytic bacteria in Polygonatum cyrtonema, and the dilution curve and coverage index reflected that the sequencing results comprehensively covered the endophytic bacterial community of Polygonatum cyrtonema. Alpha diversity analysis showed that the endophytic bacteria were highly diverse. At the level of the phylum, the dominant bacteria were Proteobacteria,Actinobacteria, Gemmatimonadetes, Firmicutes, and Acidobacteria. At the genus level, the core flora consists of Sphingomonas, Gemmatimonas, and Streptomyces. The PICRUSt gene prediction indicated that endophytic bacteria of Polygonatum cyrtonema were mainly metabolized,including energy metabolism, carbohydrate metabolism, and amino acid metabolism. Through spearman correlation analysis, the polysaccharide content associated with endophytic bacteria were obtained 19 bacterias at genus level. Among them, three genera were positively correlated and 16 genera were negatively correlated. Conclusion The diversity of endophytic bacteria in Polygonatum cyrtonema is high. There are many different kinds of bacteria, and the flora is related to polysaccharide content. The study analyzed the diversity, abundance and main genus of endophytic bacteria of Polygonatum cyrtonema, which has certain guiding significance for deep study on the internal environment of Polygonatum cyrtonema to quality.〔Keywords〕 Polygonatum cyrtonema; 16S rRNA; endophytic bacteria; diversity; community structure多花黄精为百合科(Liliaceae)黄精属(Polygonatum Mill.)多年生草本植物,以其根茎入药,在我国拥有悠久的药用历史,具有补气养阴、健脾、润肺、益肾等功效[1]。

基于16S rRNA和宏基因组高通量测序的微生物多样性研究共3篇基于16S rRNA和宏基因组高通量测序的微生物多样性研究1微生物多样性是生态学和环境科学中的一个重要研究领域,而对于微生物多样性的深入研究有助于对于地球生态系统中微生物的生态角色以及生物多样性的维持等问题进行深入探讨。
因此,本文将介绍一种用于分析微生物多样性的技术——基于16S rRNA和宏基因组高通量测序。
首先,我们来介绍一下16S rRNA这一基因。
16S rRNA是原核生物(细菌和古菌)中的16S小亚基的一部分。
这一结构在生物进化过程中相对保守,这意味着不同的物种会在16S rRNA片段中具有不同的序列差异,而这些序列差异可以用来进行物种鉴定和分类。
因此,利用16S rRNA序列可以快速准确地鉴定不同的细菌和古菌种类。
其次,宏基因组测序是指对于微生物群落中所有的基因进行高通量测序,从而可以全面地了解微生物群落的结构和功能特征。
宏基因组测序的优势在于可以同时分析多种微生物种类,包括细菌、古菌、真菌、原虫等,并且检测到微生物群落中新出现或新消失的物种,有助于对于微生物群落的动态变化进行监测。
接下来,我们来谈一谈如何利用这两种技术进行微生物多样性分析。
首先,可以从样品中提取DNA,并将其用聚合酶链式反应(PCR)扩增16S rRNA基因片段。
通过对PCR产物进行测序,可以得到一系列16S rRNA序列。
这些序列可以通过序列比对和物种分析软件进行物种鉴定和分析,从而了解微生物群落中不同物种的存在情况、数量以及相互之间的关系。
同时,宏基因组测序也可以用于微生物多样性研究。
宏基因组测序可以检测到微生物群落中每个细胞的基因组信息,并且可以同时分析多种微生物种类,从而提高其检测微生物多样性的能力。
使用宏基因组测序,可以获得微生物群落中不同基因的信息,包括种群结构、代谢途径、抗生素和毒素代谢等微生物功能信息。
这些信息有助于对于微生物群落在不同环境下的适应性以及对于环境的影响力进行深入探究。

16S rRNA基因检测技术在肠道微生态研究中的应用
黄菁华
【期刊名称】《猪业科学》
【年(卷),期】2006(23)8
【摘要】肠道微生态的研究方法主要有传统的微生物学方法和分子生物学方法.近几年来大量的分子生物学技术应用于肠道菌群的研究,取得了很大的进展,这些方法具有快速、方便、全面等优点.本文就16SrRNA主要基因检测技术(DGGE,TGGE,FISH,FQ-PCR)在肠道菌群研究中的应用做一综述.
【总页数】3页(P65-67)
【作者】黄菁华
【作者单位】即墨市兽医卫生监督检验所,山东,即墨,266200
【正文语种】中文
【中图分类】S8
【相关文献】
1.16S rRNA基因序列分析技术在细菌分类中应用的研究进展 [J], 杨霞;陈陆;王川庆
2.16S rRNA基因及多重聚合酶链式反应在新生儿败血症早期诊断中的应用研究[J], 秦桂秀;黄瑞;张莉;孟晋华;李文玲;郭慧敏;朱镭
3.16S rRNA基因检测技术在肠道微生态研究中的应用 [J], 黄菁华;张荣昌;申玉军
4.16SrRNA基因及其在水产动物分类研究中的应用 [J], 周先文;王晓清;马晓
5.16SrRNA基因检测技术在牙周病患者的口腔微环境研究的应用及诊断探索 [J], 由林;周瑞平;郭丽
因版权原因,仅展示原文概要,查看原文内容请购买。
基于16S rRNA基因的枯草芽孢杆菌PCR快速检测方法的建立李天芝;于新友;莫玲【摘要】参照GenBank中登录的枯草芽孢杆菌16S rRNA基因,设计1对引物,扩增片段大小为460bp的基因片段,该方法对枯草芽孢杆菌检测的灵敏性为1pg总DNA量,对枯草芽孢杆菌DNA的扩增结果为阳性,对照菌株扩增结果均为阴性,成功地建立了枯草芽孢杆菌PCR检测方法.该方法具有快速、灵敏度高、特异性强等优点,为枯草芽孢杆菌的分离及鉴定奠定了良好的基础.【期刊名称】《广东畜牧兽医科技》【年(卷),期】2016(041)001【总页数】4页(P30-33)【关键词】枯草芽孢杆菌;16S rRNA基因;PCR;方法;建立【作者】李天芝;于新友;莫玲【作者单位】山东绿都生物科技有限公司,山东滨州256600;;山东省滨州畜牧兽医研究院,山东滨州256600【正文语种】中文【中图分类】S816.7枯草芽抱杆菌(Bacillus Suhtilis)一种可形成芽孢的好氧革兰氏阳性菌[1],该菌具有生长条件简单、耐温度变化、快速复活、快速生长和较强分泌酶等特点。
枯草芽抱杆菌是农业部允许作为饲料添加剂的两种芽孢杆菌之一,在自然界中分布广泛,易于分离培养,对人畜无毒无害,能产生脂肽类、氨基酸和磷脂类等多种抗菌素和酶,具有广谱抗菌活性[2],可改善动物肠道菌群结构,增强免疫力,促进生长发育,提高生产性能[3]。
近年来,国内外对于枯草芽孢杆菌在饲料加工方面应用方面有量相关报道。
猪饲料中添加一定量的枯草芽孢杆菌,能增强仔猪免疫力、提高生长性能[4],蛋鸡饲料中添加枯草芽孢杆菌,即能增加种鸡的产蛋量和合格率,还能提高蛋的受精率、孵化率和健雏率[5],肉鸡饲料中添加枯草芽孢杆菌改善其肠黏膜的抗氧化和免疫功能,从而提高肉鸡的生长性能[6],枯草芽孢杆菌添加到奶牛饲料中,能显著提高奶牛产奶性能[7]。
枯草芽孢杆菌添加到兔饲料中,能显著促进幼兔肠道免疫系统发育[8]。
基于16S rRNA研究铜污染对土壤细菌群落、抗生素抗性基
因及基因转移的影响
杨潇;张娅;王宏归
【期刊名称】《扬州大学学报(农业与生命科学版)》
【年(卷),期】2024(45)1
【摘要】对经0、200、400 mg·kg^(-1)Cu^(2+)处理的3个种植池土壤分别采样,并对所有样品进行16S rRNA高通量测序。
结果表明:铜污染改变了土壤中的细菌群落结构,抗生素抗性基因(antibiotic resistance genes, ARGs)与基因水平转移(horizontal gene transfer, HGT)相关基因的丰度随Cu^(2+)浓度的升高而增加。
富集的抗性基因大多为多药耐药基因。
这表明铜胁迫会改变土壤中细菌群落结构,
促进HGT,同时使ARGs富集。
【总页数】8页(P59-66)
【作者】杨潇;张娅;王宏归
【作者单位】扬州大学环境科学与工程学院
【正文语种】中文
【中图分类】S182;S154.3
【相关文献】
1.基于16S rDNA基因文库的黄河三角洲盐生植被土壤细菌群落多样性研究
2.基
于16S rDNA高通量测序技术研究转基因作物对根际细菌群落结构的影响3.基于16S rRNA基因序列对不同土壤细菌群落的差异性分析4.基于16S rDNA测序对
茶园土壤细菌群落多样性的研究5.基于16S rRNA序列分析南糯山不同生境茶树根际土壤细菌群落分布多样性
因版权原因,仅展示原文概要,查看原文内容请购买。
第35卷第9期2015年5月生态学报ACTA ECOLOGICA SINICA Vol.35,No.9May,2015基金项目:国家重点基础研究发展规划资助项目(2013CB733502);国家自然科学基金资助项目(41371268,31300447)收稿日期:2013⁃06⁃18; 网络出版日期:2014⁃05⁃22*通讯作者Corresponding author.E⁃mail:lixz@DOI :10.5846/stxb201306181726刘驰,李家宝,芮俊鹏,安家兴,李香真.16S rRNA 基因在微生物生态学中的应用.生态学报,2015,35(9):2769⁃2788.Liu C,Li J B,Rui J P,An J X,Li X Z.The applications of the 16S rRNA gene in microbial ecology:current situation and problems.Acta Ecologica Sinica,2015,35(9):2769⁃2788.16S rRNA 基因在微生物生态学中的应用刘 驰1,2,3,李家宝1,2,芮俊鹏1,2,安家兴1,2,李香真1,2,*1中国科学院环境与应用微生物重点实验室,成都 6100412环境微生物四川省重点实验室,中国科学院成都生物研究所,成都 6100413中国科学院大学,北京 100049摘要:16S rRNA (Small subunit ribosomal RNA)基因是对原核微生物进行系统进化分类研究时最常用的分子标志物(Biomarker),广泛应用于微生物生态学研究中㊂近些年来随着高通量测序技术及数据分析方法等的不断进步,大量基于16S rRNA 基因的研究使得微生物生态学得到了快速发展,然而使用16S rRNA 基因作为分子标志物时也存在诸多问题,比如水平基因转移㊁多拷贝的异质性㊁基因扩增效率的差异㊁数据分析方法的选择等,这些问题影响了微生物群落组成和多样性分析时的准确性㊂对当前使用16S rRNA 基因分析微生物群落组成和多样性的进展情况做一总结,重点讨论当前存在的主要问题以及各种分析方法的发展,尤其是与高通量测序技术有关的实验和数据处理问题㊂关键词:16S rRNA 基因;微生物群落;多样性;高通量测序;生物信息数据处理The applications of the 16S rRNA gene in microbial ecology :current situation and problemsLIU Chi 1,2,3,LI Jiabao 1,2,RUI Junpeng 1,2,AN Jiaxing 1,2,LI Xiangzhen 1,2,*1Key Laboratory of Environmental and Applied Microbiology ,Chinese Academy of Sciences ,Chengdu 610041,China2Environmental Microbiology Key Laboratory of Sichuan Province ,Chengdu Institute of Biology ,Chinese Academy of Sciences ,Chengdu 610041,China 3University of Chinese Academy of Sciences ,Beijing 100049,China Abstract :The 16S rRNA (small subunit ribosomal RNA)gene is a universal marker for phylogenetic reconstructions to approximate the tree of life owing to its presence in all prokaryotes and its high conservation.Sequencing of 16S rRNA genes amplified directly from environmental samples is commonly used to study microbial community composition and diversity.Great advances in pyrosequencing technology and bioinformatics in recent years enable us to obtain sequence data from large⁃scale environmental samples efficiently and cost⁃effectively.However,some critical problems need to be addressed when the 16S rRNA gene is used for microbial diversity studies,such as horizontal gene transfer (HGT),intragenomic heterogeneity,PCR amplification efficiency,and sequencing data analysis.In this review,we summarize the state⁃of⁃the⁃art applications of 16S rRNA gene as a biomarker for microbial ecology studies,and introduce current pyrosequencing techniques and bioinformatics for large⁃scale data analysis.This review focuses on four aspects.(i)We introduce the structure and properties of the 16S rRNA gene,e.g.the primary and secondary structure,HGT and heterogeneities of 16S rRNA genes.Based on current available microbial genomes,multi⁃copy and intragenomic heterogeneities of 16S rRNA genes are recognized.These phenomena may seriously bias the estimations of microbial diversity in environmental samples.Some online tools and databases used for analysis of the 16S rRNA gene sequencing data are also introduced.These tools are used0772 生 态 学 报 35卷 to predict horizontal gene transfer,secondary structure,and to align and classify16S rRNA gene sequences.(ii)We introduce some16S rRNA⁃based techniques commonly used in microbial ecology studies,such as fingerprinting profiling, hybridization,microarray,and high throughput pyrosequencing methods.We compare the advantages and limitations of various methods and recommend how to use them properly based on a specific target.Different methods have different resolutions and detection limitations.Low⁃resolution profiling methods potentially miss some important information and make it difficult to detail the phylogenetic composition of an environmental sample.Pyrosequencing technique is highly recommended in the future for microbial ecology study.Several sequencing platforms,e.g.Roche454,Ion Torrent and MiSeq,are compared.(iii)We evaluate the biases that may be introduced during sample preparation and PCR procedures, e.g.DNA extraction,primer selection,PCR optimization,PCR product purification,and data analysis.Amplicon sequencing method suffers from a high level of sequencing and amplification artifacts.It is important to select OTU (operational taxonomic units)classification and chimera removing algorithms.In this case,the Uchime and Uparse are recommended for microbial amplicon pyrosequencing reads.(iv)We introduce some bioinformatics tools for pyrosequencing data analysis,such as chimera check and diversity index calculation.The most popular pipelines for pyrosequencing data analysis include RDP,QIIME and Mothur.In order to link ecological questions with microbial composition data,the methods of ecological statistics must be employed to build the relationships of microbial datasets with environmental variables.Here,we introduce some multiple statistical methods,e.g.PCA and UniFrac analysis.Based on these analyses, microbial data based on16S rRNA sequencing are linked to the environmental variables,and fundamental ecological questions are addressed.Finally,we recommend researchers to consider these problems systematically when using16S rRNA⁃based techniques in microbial ecology study.Key Words:16S rRNA gene;microbial community;microbial diversity;pyrosequencing;bioinformatics微生物是地球上数量最多和多样性最高的生物,1g土壤中仅细菌就可能有109个㊂由于大多数微生物尚不能纯培养,传统的微生物研究方法,如显微镜微形态观察㊁选择性培养基计数㊁纯菌种分离和生理生化鉴定等,在微生物多样性研究中都存在很大的局限性㊂基于非培养基础上的分子生物学方法可以使人们快速㊁系统地分析环境样品中微生物组成㊁结构和多样性,极大地促进了微生物生态学的发展㊂Zuckerkandl等首次提出使用基因序列作为分子钟来分析生物间的亲缘关系[1]Woese Fox16S rRNA的系统进化关系进行了分析,提出了著名的 三域学说”[2]㊂物,㊁基于16S rRNA信息的系统分类结果与基于全基因组信息的分类结果很相似[3]㊂随着测序技术的发展,人们可以更加快捷地获得环境样品中的16S rRNA基因序列,这些序列信息可以和数据库中的已知信息进行比对,以研究环境样品中微生物群落的特点㊂本文主要针对当前使用16S rRNA基因分析微生物群落结构和多样性的现状进行总结,重点讨论当前技术的发展状况和存在的主要问题㊂1 16S rRNA基因的特点微生物rRNA在漫长的进化过程中,由于其在碱基组成㊁核苷酸序列㊁高级结构及生物功能等方面表现出高度保守性而有微生物 化石”之称㊂保守性能够反映微生物之间的亲缘关系,为系统发育重建提供线索㊂然而rRNA的序列组成也不是完全保持恒定的,其具有一定的可变性,这种可变性能够反应出不同微生物的特征核酸序列,可以作为微生物多样性分析的分子基础㊂原核微生物的rRNA按沉降系数可以分为5S㊁16S 和23S rRNA,大小分别约为120bp㊁1540bp和2904bp左右(以Escherichia coli为例5S rRNA ,,,;相反,23S rRNA含有的核苷酸序列较长,分析较困难㊂16S rRNA占细菌总RNA量的80%以上,基因序列长短适中,其结构中既有保守区域又有变异区域,是较好的生物标志物16S rRNA 基因不同区域序列的可变性将其分为9个可变区和9个保守区(图1)㊂图1 16S rRNA 基因保守区和可变区的分布Fig.1 Conserved and variable regions in the 16S rRNA gene虽然16S rRNA 基因被广泛应用于微生物多样性分析中,然而对于某些属的菌群分辨效果较差,如Vibrio ㊁Pseudomonas 等[5]㊂人们一般设定16S rRNA 基因序列相似性³97%的原核生物为同一个种,但很多不同种的微生物其16S rRNA 基因序列相似性却高于97%㊂另外,许多细菌的16S rRNA 基因是多拷贝的,而且各拷贝的序列组成存在一些差异;16S rRNA 基因也存在着水平转移问题,这些都直接影响着对原核微生物群落结构和多样性的分析㊂1.1 基因水平转移(HGT)人们以前曾普遍认为16S rRNA 基因不存在水平转移现象,然而一些研究发现,在许多细菌中16S rRNA 基因都出现了水平转移[6⁃7]㊂在对Pseudomonas 属的研究中发现,有些种的V1区和V3区中的48.3%和41.6%的16S rRNA 基因序列可能是从另一个距离较远的种通过基因水平转移得到的,因此,应该避免使用这些可变区对Pseudomonas 进行种间分类[7]㊂Choi 等在对Pfam 数据库蛋白结构域分析时发现已有的微生物可能发生过基因水平转移的占1.1% 9.7%,其中多于一个蛋白结构域基因发生水平转移的古菌占一半以上,而细菌占30% 50%,真核生物不到10%,这说明原核微生物发生基因水平转移的频率远远大于高等生物,他们也通过对比发现HGT 对于群落SSU (small subunit)rRNA 基因整体的系统发育分析影响不大[8]㊂Garcia⁃Valle 等建立了基因水平转移数据库(HGT⁃DB),收录了多种微生物发生基因水平转移的数据和相关证据[9]㊂1.2 16S rRNA 基因的多拷贝及异质性很多原核微生物基因组中rRNA 基因是多拷贝的,同一菌种中不同拷贝的基因序列也可能存在差异(异质性)[10]㊂Větrovsky ′等研究了1690种已知的细菌基因组,发现只有15%的基因组中16S rRNA 基因是单拷贝的,21%的基因组含有3 7个拷贝,最大的拷贝数是15(Brevibacillus brevis NBRC 100599和Photobacteriumprofundum SS9),2.4%的基因组中16S rRNA 基因具有1%以上的序列差异[11]㊂在此之前,Pei 等[12]通过对数据库中的rRNA 序列进行分析时也发现,425个种都具有2 15个不等的rRNA 基因拷贝数(2.22±0.81),如古菌一般含2 4个,氨氧化细菌含1 2个,固氮菌一般含1 3个;235个基因组中16S rRNA 基因序列异质性变化在0.06% 20.38%之间,例如古菌Haloarcula marismortui 基因组含有的两种16S rRNA 基因序列差异性达到了5%㊂多拷贝的存在给微生物群落的定量分析带来诸多问题,使群落的多样性估计和结构分析存在较大的偏差[13]㊂Větrovsky ′等使用高通量测序法分析土壤细菌时发现具有低拷贝16S rRNA 基因的类群如Acidobacteria 的丰度会被低估,而具有高拷贝16S rRNA 基因的类群如Gammaproteobacteria 的丰度会被高估㊂在某些特定情况下,对于拷贝数多或异质性大的菌群可以用其它替代性的单拷贝基因来分析其多样性或进化关系,如GroEL 伴侣蛋白,RNA 聚合酶β亚基(rpoB ),DNA 回旋酶β亚基(gyrB )和热休克蛋白(dnaK )等[14⁃15]㊂也有人使用hsp 60进行Enterobacter 的系统进化和分类研究[16]㊂异质性问题对16S rRNA 基因高通量测序研究的影响还需要进一步系统性评估㊂1772 9期 刘驰 等:16S rRNA 基因在微生物生态学中的应用 2772 生 态 学 报 35卷 1.3 16S rRNA序列的二级结构不论种间的16S rRNA基因序列差异有多大,在进化过程中16S rRNA都保留了相似的二级结构,与Escherichia coli的二级结构相似性很高[17]㊂Thermoanaerobacter tengcongensis含有4个16S rRNA基因,这些基因的一级结构差别达到了6.7%,但二级结构却是很保守的[12]㊂研究者通过元基因组学方法发现其它细菌rRNA序列与Escherichia coli的16S rRNA序列相似性为80.9% 99.0%时,它们的rRNA操纵子可以通用,只不过代时将会增加,这个研究表明保持二级结构是维持rRNA功能的重要前提㊂虽然HGT对微生物群落多样性的分析结果影响较小,但如果进行研究的某些微生物类群中16S rRNA基因发生HGT可能性较大,要精确构建进化树则需根据已有的实验经验综合进行分析㊂RNA分子具有降解速度快且难以结晶的特点使得难于通过X射线晶体衍射和NMR(Nuclear Magnetic Resonance)等方法提高对RNA分子空间结构的认识,因此利用各种计算方法从理论上分析rRNA的空间结构是目前主要的方法㊂原核微生物16S rRNA的二级结构相对于一级结构更加保守,因而研究二级结构对于系统进化分类很有意义[18],尤其是对于种属的鉴定[19]㊂很多序列比对分析软件都考虑了二级结构,如RDP aligner㊁ARB(http://www.arb⁃home.de/)㊁SSU⁃ALIGN(/software/ssu⁃align/)等[20⁃21],使系统进化分类结果更加快速准确㊂rRNA二级结构分析编辑可使用ARB[22]和jphydit(http://plaza.snu.ac. kr/ jchun/jphydit/index.php)等㊂RNA二级结构的预测主要包括2种方法:最小自由能算法和序列比较分析方法㊂当前核酸数据库日益庞大,数据量呈指数性地增长,在16S rRNA一级结构的基础上进行二级结构分析对序列比对㊁OTU(operational taxonomic units)分类就变得更加重要[23⁃24],因此应该加强二级结构分析预测算法及软件的研究㊂1.4 16S rRNA基因序列分析数据库研究者们已经建立了一些专门针对于16S rRNA的数据库,储存16S rRNA基因序列,通过互联网对新测定的序列进行比对分析,比较著名的数据库有SILVA(http://www.arb⁃silva.de)[25]㊁RDP(http://rdp.cme.msu. edu/)[21]和Greengenes(/)[26],人们可以从这些数据库中获取高质量的16S rRNA基因序列信息㊂rrnDB(/rrndb)数据库专门用于记录细菌和古菌基因组中rRNA和tRNA基因的拷贝数[27]㊂在NCBI㊁DDBJ和EMBL数据库中也有许多16S rRNA基因全长和短序列,如NCBI Genbank中有几十万条大于1000bp的人体微生物群落的16S rRNA序列㊂某些领域也建立了一些专业的数据库,如病原微生物16SpathDB(http://147.8.74.24/16SpathDB)数据库,可用于鉴定对临床重要的细菌16S rRNA序列[28];CORE[29]是人体口腔微生物方面的专业数据库㊂Comparative RNA Web Site(http://www.rna. /)是分析rRNA二级结构的数据库㊂EzTaxon⁃e收集了已培养或未培养微生物的16S rRNA序列信息,常用于鉴定新分离的菌株[30]㊂probeBase可用来检索与16S rRNA有关的探针[31]㊂2 16S rRNA基因在研究微生物群落中的应用常规的微生物分子生物学研究方法如分子杂交㊁SSCP㊁DGGE㊁RFLP㊁ERIC⁃PCR㊁FISH及克隆文库法等相对简单,易于实验操作,但得到的信息量有限,分辨率普遍较低㊂近些年来发展起来的高通量测序[32⁃33]和基因芯片技术[34⁃35]强有力地推进了微生物生态学的研究㊂尽管每种方法都有各自的局限性,但这些技术的应用使人们对环境中的微生物群落有了更深入的认识[35⁃36]㊂图2勾画出了常用的利用16S rRNA基因分析微生物群落结构和多样性的基本流程㊂2.1 指纹图谱技术基于16S rRNA基因的指纹图谱技术如DGGE㊁TGGE㊁SSCP㊁ARDRA和T⁃RFLP等被广泛用于微生物群落的研究中[37]㊂由于指纹图谱技术的操作相对较简单,在环境微生物动态监测和分析等方面得到了广泛应用[38⁃39]㊂表1列出了常见的指纹图谱分析方法的原理和优缺点,并与多基因序列分析(MLSA)进行比较㊂图2 基于16S rRNA 基因的微生物群落多样性分析的主要流程Fig.2 Pipelines for microbial diversity analysis based on the 16S rRNA gene表1 各种rRNA 分析技术的原理和比较Table 1 Comparisons of fingerprinting profiling techniques方法Method原理Principle 特点Characteristic 参考文献References 核糖体分型(Ribotyping)利用限制性内切酶对整个基因组进行酶切,用标记的16S rRNA 基因探针与之杂交,检测相应的RNA 操纵子的位置只与限制性酶切位点的变化相关,需进行杂交试验,较费时[40]扩增性rDNA 限制性酶切片段分析(ARDRA)对16S rRNA 基因扩增或克隆产物进行限制性酶切片段长度多态性分析分辨率高,与限制性位点有关,费时[41]末端限制性片段长度多态性分析(T⁃RFLP)通过PCR 技术㊁DNA 限制性酶切技术和荧光标记技术得到末端带有荧光标记的限制性多态性片段方便快捷,灵敏度高,并只与末端限制性片段的大小有关[39]16S 23S 内部转录间隔区(ITS)分型ITS 片段在进化过程中产生更多的变异,即使是亲缘关系非常接近的2个种都能在ITS 序列上表现出差异比16S rRNA 基因序列具有更大的变异,对亲缘性更近的种属分型更有用[42]核糖体ITS 区自动分型(ARISA)利用荧光标记微生物核糖体基因间隔区(ITS)对差异进行分析分辨率较高[43]限制性片段长度多态性分析(RFLP)整个rRNA 基因的限制性片段长度多态性分析灵敏度高,操作要求高,分析较复杂[44]变性梯度凝胶电泳(DGGE)DNA 在不同浓度的变性剂中解链行为不同,导致电泳迁移率发生变化,从而将片段大小相同而碱基组成不同的DNA 片段分开检测片段在500bp 以内为宜,中等分辨率,只能检测优势菌群[45]温度梯度凝胶电泳(TGGE)DNA 在不同温度梯度环境中解链行为的不同会导致电泳迁移率发生变化大片段DNA 由于Tm 值较高,检测难度较大[46]单链构象多态性(SSCP)单链DNA 的构象差异导致其在凝胶电泳中的迁移率变化分辨率中等,检测片段小于400bp 最为理想[47]多基因序列分析(MLSA)通常对几个长度为500bp 左右的目的基因进行测序,用于进化分析能有效地在种水平上鉴定菌株,还可用于生态型的划分[48] ARDRA:amplified ribosomal DNA restriction analysis;T⁃RFLP:terminal restriction fragment length polymorphism;ITS:internally transcribedspacer;ARISA:Automated ribosomal intergenic spacer analysis;RFLP:Restriction fragment length polymorphism;DGGE:Denaturing gradient gel electrophoresis;TGGE:Temperature gradient gel electrophoresis;SSCP:Single⁃strand conformation polymorphism;MLSA:Multilocus sequence analysis 3772 9期 刘驰 等:16S rRNA 基因在微生物生态学中的应用 4772 生 态 学 报 35卷 整体来看,指纹图谱技术通量和分辨率比较低,可以用来对样品进行初步的筛选或评估,例如有时为了减少测序量,可以先进行ARDRA或其它方法分型,然后根据分型结果对有代表性的样品再进一步研究㊂某些情况下如果仅使用16S rRNA的信息,分辨效果可能不是太好,可使用其它一些分子分析方法作为补充㊂相比16S rRNA和23S rRNA来说,ITS序列在进化过程中由于面临着较小的选择压力故存在较大的变异,因此ITS 的长度和序列的多态性能用来区分不同种的原核生物[42]㊂在种的水平上,MLSA也是鉴定菌株非常好的方法[48],可利用16S rRNA基因序列和各种指纹技术将未知菌株聚类到一个科或属里,然后根据这一组菌株的特点选择某些代表性的标记基因进行MLSA分析,从而进行种㊁亚种以及生态型的划分㊂有时也可参考数据库MLST()或PubMLST()进行一些病原微生物的多位点序列分型方面的信息查询㊂某些微生物之间的16S rRNA基因序列几乎相同,但是DNA杂交值却明显低于70%,因此它们代表着不同的种㊂16S rRNA基因同源性低于97%的菌株间其基因组DNA的相似性均低于60%㊂DNA⁃DNA杂交(DDH,whole genomic DNA⁃DNA hybridization)实验操作复杂,不适合进行较大范围的系统分类研究,因此16S rRNA序列分析的结果可以说明是否有必要进行DDH实验㊂随着高质量序列数据的快速增长,ANI(average nucleotide identity)作为一种很精确且方便使用的分类方法,应用越来越广泛[49]㊂2.2 荧光原位杂交技术(FISH)FISH技术是一种不依赖PCR的分子技术,它结合了分子生物学的精确性和显微镜的可视性,可原位监测和鉴定样品中不同的微生物个体,已经广泛应用于微生物定量分析和群落结构分析[50⁃51]㊂FISH的原理是根据待测微生物的16S rRNA基因中的保守序列,设计相应的特异性寡核苷酸探针,并进行荧光标记,通过探针与环境基因组中的DNA分子进行杂交,检测该特异微生物种群的存在和丰度㊂利用分子遗传上保守性不同的特异序列,可以在不同的分类水平(如属㊁种等)上进行检测㊂当前的荧光探针安全且具有很好的分辨力,如果使用具有不同激发和散射波长的荧光染料标记探针,可同时检测多个靶序列[52⁃53]㊂FISH技术与其它方法结合能够更好地发挥优势,这些技术包括microcolony⁃FISH㊁DVC⁃FISH㊁in situ PCR⁃FISH㊁CPRINS⁃FISH㊁CARD⁃FISH㊁MAR⁃FISH等[53⁃54],其中CARD⁃FISH技术很好地解决了目标微生物数量较少时信号检测灵敏度低的问题[55]㊂Behrens等利用EL⁃FISH与NanoSIMS技术进行了微生物群落中单细胞进化分类和代谢活性的研究[56]㊂FISH技术成功应用的关键在于设计和获得具有高灵敏性和专一性的寡核苷酸探针以减少干扰,可使用ARB软件包结合probeBase数据库等多种方法进行探针分析和设计㊂综合来看,应用FISH技术不仅能研究微生物群落的结构特征和空间分布,还可以跟踪微生物种群的动态变化,此外利用mRNA等作为目标分子还可以进行群落代谢方面的研究㊂2.3 实时定量PCR(qRT⁃PCR)qRT⁃PCR是通过对PCR扩增反应中每一个循环产物荧光信号的实时检测实现对起始模板定量的分析,具有特异性强㊁重复性好㊁准确快速等优点㊂通常使用的荧光化学方法有TaqMan荧光探针法和SYBR荧光染料法等㊂许多因素都会影响qRT⁃PCR反应在定量上的准确性,如细胞裂解效率㊁抑制剂的去除程度㊁SYBR GreenⅠ的浓度㊁同源和异源DNA背景㊁操作流程设计㊁目标基因的选择以及PCR产物的长度等,因此在操作时需要优化条件,最大化地减小误差[57⁃60]㊂各种qRT⁃PCR方法都有其自身的优缺点,操作时需要根据实验设计特点选择㊂对于特定类群微生物也可使用一些功能基因代替16S rRNA基因进行定量,如利用mcr基因定量产甲烷菌[58]㊂在绝对定量时,要用到标准样品,可以用带有特定基因的质粒,或者利用直接从环境样品中扩增的PCR产物做标准样品㊂各种实验操作因素(不同反应批次的差异㊁加液误差等)对qRT⁃PCR结果影响很大,因此在定量多个环境样品时,最好能用96或384孔的反应板一次完成㊂qRT⁃PCR既可以对微生物群落的总体进行定量分析,也可以对具体的种属进行定量研究㊂某些种属的16S rRNA基因分辨率较低,可使用一些功能基因进行定量,如使用特异的invA基因研究Salmonella属㊂Liu 等设计了一种新的qRT⁃PCR方法用于定量分析群落16S rRNA基因的丰度,并分析了rRNA基因拷贝数等对定量的影响[13]㊂qRT⁃PCR与其它技术相结合使用可更好地研究群落的结构和多样性,如指纹图谱技术㊁测序技术等[61]㊂2.4 基因芯片技术基因芯片技术的突出特点在于其高度的并行性㊁多样化㊁微型化和自动化㊂基因芯片的测定结果与高通量测序[62]㊁qRT⁃PCR[63]的结果有很好的一致性㊂根据已知的基因序列信息,研究者们设计出了许多基因芯片进行微生物群落结构和功能的研究,包括基因组芯片㊁功能基因芯片和系统进化芯片等,主要用于评估多样性和鉴定病原菌等㊂其中系统进化芯片(PhyloChip)是含有与SSU RNA基因序列互补的寡核苷酸探针的阵列,适合于微生物群落结构组成和动态变化分析㊂PhyloChip已应用于研究人的肠道㊁口腔以及水体㊁土壤和植物根系等微生物群落[64⁃66]㊂利用微生物的功能基因作为探针开发出的功能基因芯片 GeoChip[67],已广泛应用于检测各种环境样品中微生物群落的功能基因[68⁃69]㊂基因芯片的信号扫描及数据处理有许多商业软件可供使用,一些开源工具如Bioconductor平台(/)[70]和R语言(http://www.r⁃/)也常用于基因芯片的数据分析中㊂在定性分析方面,基因芯片是根据已知的基因序列信息制作的,因此无法检测到未知的基因信息,也可能检测不到那些丰度较低的基因㊂检测范围较广的基因芯片由于探针数目太多易产生假阳性,设计特异性探针能够克服这类问题,比如近些年来开发出专门用于检测人体微生物相关菌群的HITChip[71]及用于检测口腔微生物的OC Chip[72]等㊂在定量分析方面,由于某些菌rRNA基因操纵子是多拷贝的,而使测定的信号总量代表的是16S rRNA基因的总拷贝数,只有使用改进的算法才能更精确地进行定量分析[73]㊂与高通量测序相比,基因芯片方法实验费用低,数据分析过程相对简单,有时在研究特定的菌群和功能基因时,基因芯片技术是一个较好的选择㊂而高通量测序是一个 开放系统”,可以探测到环境中新的基因信息,信息更加系统㊁丰富,但需要利用生物信息学方法和生物化学知识对序列数据进行深入挖掘㊂2.5 高通量测序技术要全面系统地分析微生物的多样性,首先要获取16S rRNA基因序列,如何快速准确地获取大量的序列信息就显得很重要㊂传统的Sanger测序方法操作复杂㊁通量低,难于解决这个问题㊂2005年,美国454Life Science公司首先推出了革命性的基于焦磷酸测序法的高通量测序系统,将第二代测序技术推向了市场㊂此后,Illumina公司和ABI公司相继推出了Hiseq和SOLiD等测序平台㊂现在利用高通量测序技术研究环境微生物多样性的报道越来越多,这些系统的主要优点在于通量高,而且利用Barcoded PCR技术使得在一次运行中能同时进行多样本的大规模测序,使测序成本大大降低㊂基于单分子测序原理的第三代测序技术也在迅速发展,Helicos Biosciences公司推出了单分子DNA测序仪tSMS Performance,PacBio与ZSGenetics公司也在大力研发新一代测序仪[74],第三代测序技术的进步将使测序更加快速准确,且能大大增加读长,而且可以直接对RNA测序㊂表2总结了几种最常用的高通量测序仪的主要技术参数㊂表2 目前常用的高通量测序平台的主要技术参数Table2 Main technical specifications of several pyrosequencing platform测序仪类型Platform运行时间Run time读长(bp)Length(bp)运行通量Throughput错误类型Error typeRoche454GS FLX Titanium XL+ 454GS FLX Titanium XLR7023h10h<1000<600≤700Mb≤450MbIndelIndelIllumina GAIIx HiSeq2000 MiSeq 14d11d65h2×2×2×300≤95Gb≤600Gb13.2 15GbSubstitutionSubstitutionSubstitutionLifeSOLiD5500xl8h75×35PE<300Gb(nanobeads)A⁃T bias Ion TorrentPGM316chip v2 4.9h400≤1Gb Indel 5772 9期 刘驰 等:16S rRNA基因在微生物生态学中的应用 6772 生 态 学 报 35卷 基于测序技术进行微生物多样性研究的方法主要有两种:一种是基于16S rRNA基因的扩增子测序,另一种是基于环境基因组DNA的宏基因组测序,两种方法各有优缺点[75⁃76]㊂相比之下,基于16S rRNA基因的高通量测序技术更加常用,并且消除了克隆问题,可以综合研究各个可变区,分析方法也相对成熟,不过在具体的实验分析中也存在着一些问题需要解决,例如PCR扩增的偏差㊁扩增错误㊁测序错误等都可能影响群落α⁃多样性的准确估计[77⁃79]㊂在16S rRNA基因扩增子测序中,Roche GS FLX+测序平台理论读长最高可以达到1000bp,一块PTP板可获得超过80万条序列,而Illumina测序平台具有通量高的优点[80],使得Miseq和Hiseq系列测序仪广泛用于微生物生态学研究中㊂宏基因组测序无需PCR扩增,既可以研究微生物的群落组成和多样性,也可以反映群落的很多代谢特征,但数据分析相对复杂㊂3 16S rRNA基因实验操作过程中的问题3.1 核酸提取环境基因组总DNA的提取效率影响着对微生物群落组成的准确鉴定和定量分析㊂核酸提取主要分为细胞破碎㊁核酸沉淀以及后续纯化三个环节㊂环境样品DNA提取过程中,不完全的细胞裂解㊁基质对DNA的吸附㊁酶抑制剂的共提取以及提取过程中DNA或RNA的降解是主要的一些问题[81],尤其是土壤样品,提取DNA或RNA过程中往往浸提出腐殖酸物质,抑制PCR或反转录反应㊂商业化的试剂盒(比如MP公司的FastDNA Kit for Soil㊁Mo Bio公司的PowerSoil DNA Isolation Kit等)能够部分解决此问题,不过价格较贵㊂其它成本较低的方法包括SDS裂解法㊁酶解法㊁玻璃珠破碎法及超声波破碎法等,这些方法提取的DNA往往需要后续再纯化以满足实验需要㊂对于抑制物含量较高的土壤DNA,可以用水稀释或利用CTAB方法进一步纯化后再扩增㊂研究表明,初步提取的DNA利用CTAB或/和PVPP可以有效去除大部分抑制剂或杂质[82]㊂针对不同的环境样品,应该根据文献报道的相关结果对DNA提取方法进行优化㊂Canto等在研究污泥微生物时使用不同的细胞直接裂解方法,获得了一种实用有效的DNA提取方法[83];Kuczynski等建议在研究复杂微生物群落时应使用多种方法进行细胞裂解[84];Flores等研究了一种新的直接PCR法以简化DNA提取步骤[85]㊂对于粘性土壤,可以加入skim milk以提高DNA的提取效率[86]㊂用试剂盒提取的DNA,进行qRT⁃PCR实验时的平行性往往优于自配试剂的提取方法㊂在进行样品总RNA提取过程中,要特别注意污染和降解问题[87],一些RNA酶抑制剂可以用于样品运输过程中的保存,以防止RNA降解,如RNA later和Methanol/ HEPES溶液[88]等㊂必须注意的是,要使结果分析重复性和精确性较高,在提取以及后面的操作过程中都要严格控制污染问题,因为相比在测序和分析过程中引入的偏差,实验污染等问题可能对结果影响更大㊂3.2 引物的选择16S rRNA基因不同区域的保守度是不一样的,因此扩增区域的选择会影响多样性的分析结果[89]㊂研究者们从各个方面对引物的选择问题进行了分析,主要有引物长度㊁扩增长度选择㊁引物覆盖度等㊂Cai等发现污泥微生物多样性分析与扩增区域有很大的关系,测序深度会影响低丰度细菌的鉴定[90]㊂利用27F/338R (V1 V2)引物,人们发现Verrucomicrobia类群在研究的土壤中数量相对较少,但改用515F/806R(V4)引物后,发现土壤中该类群的丰度变大[91]㊂理想的引物是能够尽量多地将环境样品中的基因扩增出来,同时又要能够区分出不同的种属㊂为了提高对环境样品DNA扩增的覆盖度,可以设计简并性通用引物,即使这样,也不能覆盖所有的菌群[92],而且,增加位点的非特异性可能会增大扩增偏差的概率㊂表3[93⁃101]列出了一些常用的16S rRNA基因的引物,不同的引物对不同种属的覆盖度不一样,某些引物对细菌有很高的覆盖率,某些专门针对古菌,还有些是细菌和古菌通用的引物㊂有的学者建议细菌与古菌群落应该分别用专用引物进行分析[102],而其他一些学者则直接使用细菌与古菌通用引物[95⁃96]㊂因此,在实验设计时需要考虑到实验目的而有针对性地进行选择,对不同的通用引物的覆盖能力要有个大致的了解㊂利用454GS FLX平台,测序长度可以达到800bp,然而PCR产物短时,得到的序列数量多㊁质量好,400bp左右的扩增子进行测序目前看来引物的选择很大程度上还是要根。
第41卷 第4期河南农业大学学报V o.l 41 N o .42007年8月Journa l of H enan A gricultural Un iversityAug .2007收稿日期:2007-03-20基金项目:河南省自然科学基金项目(0511030400)作者简介:黄进勇(1969-),男,河南周口人,副教授,博士,从事农业环境生物学研究.文章编号:1000-2340(2007)04-0396-05麦田土壤细菌群落16S r DNA V3片段PCR 产物的DGGE 分析黄进勇,岳彩鹏,周 伟(郑州大学生物工程系,河南郑州450001)摘要:以郑州市郊冬小麦农田土壤为研究对象,利用改进的土壤DNA 提取方法提取土壤微生物基因组,并采用降落式PCR 和DGGE 电泳技术对细菌16S r DNA V 3区进行扩增和产物分离,分析了小麦4个生育时期3个土层深度的细菌群落变化.结果表明,麦田土壤细菌多样性极高,小麦整个生育期土壤中一直存在数种优势菌群.但各时期都有新的不同的细菌出现,整体表现为随着小麦的生长多样性逐渐增加,收获期减少.表层土壤各生育时期菌群数量变化较大,而深层土壤菌群数量变化较小.关键词:土壤;冬小麦;细菌群落;DGGE 中图分类号:S 512.1 文献标识码:AAnalysis of Bacteriu m Co mmunity i n W i nter -W heat Soil Usi ng DGGEof 16S r DNA V3Frag m ent PCR ProductsHUANG Ji n -yong ,YUE Ca-i peng ,Z HOU W e i(Depart m ent of B i o -Eng i n eering ,Zhengzhou University ,Zhengzhou 450001,China)Abst ract :U si n g i m proved extraction m ethod o f soil DNA and so il fro m the w i n ter wheat fields inZhengzhou suburb as experi m ent subjec,t w e ex trated the DNA geno m e fro m so ilm icrob ials .In th is paper ,the diversity of bacterial co mm un ity w as stud ied by a mp lify i n g the bacterial 16S r DNA V3frag -m ent w ith touch -do w n PCR m ethod ,and separating t h e products by denaturi n g g radien t gel electropho -resis (DGGE).So il sa m ples w ere taken at three d ifferent depths at four d ifferent stages .The resu lt sho w ed tha,t the diversity of so il bacteria i s higher in wheat fields .W ith the gro w t h ofw hea,t the d-i versity is i n creasi n g gradually ,and decreasi n g i n harvest per i o d .There are a fe w stable and do m i n ant bacteria co mm un itiy i n t h e w ho le wheat gro w th period ,but so m e ne w different bacteria appeare at every period .The nu m ber o f bacteria co mmuniti y i n top so il changesm ore greatly t h an i n deep so il i n differ -ent period .K ey w ords :so i;l w i n ter whea;t bacteri u m co mmun ities ;denaturi n g grad i e nt ge l electrophoresis (DGGE ) 冬小麦-夏玉米种植模式是中国北方地区广泛采用的集约化种植方式,通过对该模式下土壤细菌群落多样性的变化研究,将为该地区农业生产模式的改进提供理论支持.传统的微生物多样性研究以纯培养技术为主,但实验室能够分离培养出的微生物只占其中的0.01%~10.0%,不能很好的反映微生物的实际存在状态[1].近年来发展起来的变性梯度凝胶电泳(Dena t u ring grad ient gel e lectro -phoresis ,DGGE )技术则是基于16S r DNA 保守性的DNA 指纹技术,在不需要纯培养的条件下能够对微生物的复杂群落进行有效分析[2].作者采用PCR-DGGE 技术对小麦不同生育期及不同土层深第4期黄进勇等:麦田土壤细菌群落16S rDNA V 3片段PCR 产物的DGG E 分析397度的细菌多样性进行了研究,以期为调控农业生产方式和进行合理的生产管理提供科学依据.1 材料与方法1.1 供试土壤样品土壤样品取自郑州市郊水肥管理一致且肥力相对均匀、常年种植冬小麦-夏玉米的集约化种植农田,在固定位置采取5点取样法取样,每个样点取3个层次的土壤样品,分别为0~10,10~20,20~30c m.取样时间分别为小麦苗期、拔节期、扬花期及收获后4个时期.另取田边土壤作为空白对照.1.2 研究方法1.2.1 土壤全基因组提取 为减少土壤腐殖质等杂物干扰,对ZHOU [3]所报道的土壤基因组提取方法进行了改进.具体提取方法为:将5g 土壤加入1315mL TENC (50mm o l #L -1Tris -H C ;l 20mm o l #L -1EDTA;100mm ol #L -1磷酸钠;1%CTAB ),并加入25L L 蛋白酶K,混匀后置于37e 摇床225r #m i n -1震荡30m in ,然后加入1.5mL SDS 后混匀,65e 水浴2h (每隔10~20m in 摇动1次),6000r #m i n -1离心15m in ,转移上清液至新的离心管中;在原离心管中加入9mL TENC 及1mLSDS,混匀后放入水浴中1h ,6000r #m i n -1离心15m in ,取上清并,合并上次上清,加入等体积的氯仿/异戊醇,充分混匀后10000r #m in -1离心15m in ,吸取上清至新离心管中,等体积加入异丙醇,混匀并放入冰箱中4e 过夜,10000r #m i n -1离心15m in ,弃上清并加入2mL 预冷的70%乙醇洗涤2次,空气干燥后加入500L L TE 溶解.在018%的琼脂糖凝胶中对获得的总DNA 进行电泳,以检测是否得到较为完整的总DNA.1.2.2 基因组DNA 的纯化及16S r DNA 可变区的PC R 扩增 采用宝生物公司的凝胶回收试剂盒对提取所得的基因组DNA 进行纯化,得到23kb 左右的DNA 片段.然后以此DNA 作为模板,对16S r DNA V3可变区进行PCR 扩增.扩增引物为通用的F338GC 和R518(对应于大多数细菌和古细菌的16Sr RNA 基因的特异性V3区),其中正向引物5.端连接有GC 夹板,以提高在后期DGGE 电泳时的解链范围.引物的序列分别为F338GC :5.-CGC -CCGCC GCGCGCGGCGGGCGGGGCGGGGGCACGG-GGGGCCTACGGGAGGCAGCAG-3.;R518:5.-AT-TACC GCGGCTGCTGG-3..扩增反应体系为50L L:5L L 10@PCRbuffer (M g 2+Plus),4L L dNTPs(各215mm o l #L -1),1L L 各种引物(10L m o l #L -1),1L L 模板,0.5L LTaqDNA 聚合酶(5U #L L -1).扩增条件采用梯度PCR 策略:94e 预变性5m in ;前24个循环为94e 45m in ,65~53e 1m i n (每循环降低0.5e ),72e 45m in ;后11个循环为94e 45m i n ,53e 1m i n ,72e 45m i n ;最后延伸5m i n .1.2.3 16S r D NA V3区PCR 产物的变性梯度凝胶电泳 试验采用B io -Rad 公司的D-Code syste m 基因突变检测系统对PCR 反应产物进行分离,10%聚丙烯酰胺凝胶,变性剂梯度为30%~70%(100%变性剂为7m o l #L -1尿素和40%去离子甲酰胺混合物),PCR 产物加样量30L L ,在60e ,1@TAE ,130V 条件下电泳5h ,电泳胶片利用改进的B assa m 银染方法[4]进行染色.1.2.4 电泳图谱的统计学分析 观察各个土壤样品的PCR 产物经变性梯度凝胶电泳(DGGE)分离后的电泳图谱照片,采用天能G I S 凝胶成像分析系统,对各土壤样品的电泳条带的多少及密度来进行分析,并绘制泳道间遗传图,以初步统计多样性指标数据.2 结果与分析2.1 基因组总DNA 提取效果通常提取的土壤微生物基因组DNA 的大小应该在15~23kb 范围内,所以采用K -H in d ⅢM arker 作为标准分子量DNA,对获得的总DNA 在0.8%的琼脂糖凝胶中进行电泳,结果显示(图1),所采取的改进方法所得总DNA 在23kb 出现1条较亮条带,表明该法得到较为完整的总DNA.M :K -H i n d ⅢM arker ;B :无土壤的空白对照;1,2:改进方法的2个重复.M:K -H in d Ⅲm ark er ;B :the b l ank ;1,2:The res u lt of the i m p roved m ethod图1 改进方法提取土壤微生物基因组DNA 效果的电泳图Fig .1 E lectrophoretogra m of DNA extraction fro m thesoil u si ng i mproved extrac ti on m e thod398 河 南 农 业 大 学 学 报第41卷2.2 基因组DNA 的纯化及PCR 扩增结果结果表明(图2),通过该PCR 程序,扩增得到大约260bp 左右的产物,与理论值相符.说明该PC R 程序适合用于16S r DNA 的扩增,并且能够得到较好的产物.2.3 目的片段的DGGE 分离扩增得到的不同时期不同深度土壤样品16S r D NA V3可变区,通过DGGE 电泳进行分离,DGGE 条带数与土样中细菌种群的数量相关,而条带的亮度则反映该种细菌数量的多少.电泳图谱显示(图3,图中1~12各泳道代表的内容同图1),DGGE 电泳分离出较多的条带,显示农田土壤中细菌的多样性相当丰富.从图3可看出,各个时期及层次的样品具有较高的一致性,但是,随着时期的变迁和取样深度的变化,微生物的多样性又随之发生一定的变化.在所有时期的不同层次的土壤中,编号分别为a ,b ,c 的3条分离带一直存在,虽然亮度有所变化,但亮度均比较高,表明这3种细菌是该类型土壤中的基本种群,在数量上也具有优势;而1,2,3号条带除了在个别样品没有出现外,存在大部分样品中,暗示了这些细菌受小麦生育期及土壤深度的影响较小,具有一定的稳定性;4号条带从小麦苗期30c m 土样中开始出现,一直持续到收获后期,且亮度较1~3:空白10,20,30c m 土壤;4~6:苗期10,20,30c m 土壤;7~9拔节期10,20,30c m 土壤;10~12:扬花期10,20,30c m 土壤;13~15:收获后10,20,30c m 土壤.1~3:t he b l ank s oil of 10,20,30c m;4~6:t he seed i ng s oil of 10,20,30c m;the j oi n ti ng stage soil of 10,20,30c m;the an t hesis st age s o il of 10,20,30c m;the harvest stage s o il of 10,20,30c m图2 土壤16S rDNA 基因V3区扩增片断F ig .2 PCR a m plified frag m en t 16S rDNA (V3)gene of d ifferen t s o il sa m p l e s高,则说明该种细菌与小麦的生长发育相关;5,6号条带只在苗期30c m,扬花期30c m 以及整个拔节期出现,表明其与小麦拔节期前后的生长相关;而7号条带只在苗期与拔节期出现.进行分离的15个样品的泳带均有所变化,说明随着生育期的变迁与土壤深度的变化,该地区土壤中的细菌群落不断发生变化,具有相当丰富的多样性.从条带的丰富度来看,随着时间变化一些条带消失,一些新的条带产生,但条带总数则不断增加,直到后期才又有所减少,说明土壤中不同种类的细菌随小麦的发育彼此消长,但种类不断增加.2.4 电泳图谱的统计学分析绘制了不同时期及不同层次土壤之间的遗传簇关系(图4).该图通过各泳道所代表遗传簇的异同,表示各样品之间的基因多样性及亲缘关系.由分析结果可知,所有样品之间DGGE 谱带的相似性较差,表现出了较好的多样性.这种多样性同时受作物生育时期和土壤层次的影响.条带1,2,3归于一簇,说明同一时期不同土层深度的细菌变化不大,条带5,6以及条带8,9各归于一簇,说明拔节期和苗期土壤细菌受小麦生长的影响比深度的变化对其多样性的影响要大.第4期黄进勇等:麦田土壤细菌群落16S rDNA V3片段PCR产物的DGG E分析399图3V3区扩增片断的DGGE分析结果Fig.3DGGE analytical resu lt of V3frag m ents3讨论由于土壤成分复杂,常规提取总DNA的方法难以去除腐殖酸,而微量的腐殖酸即可抑制后期的分子生物学实验,因此建立高效、可靠的微生物总DNA提取方法是进行微生物分子生态学研究的基础.我们在前人工作的的基础上,对ZHOU[3]所报道的土壤基因组提取方法进行了改进.主要是降低了原有DNA提取缓冲液的浓度,减少了蛋白酶K的用量,并适当延长了裂解时间.研究证明,该方法适于郑州市郊区小麦土壤微生物高效、高质的基因组DNA提取,以用于遗传多样性研究.通过DGGE分析发现,麦田土壤细菌多样性极高,随着时间的迁移,土壤中一直存在数种菌群,而且泳带亮度较大.由于DGGE通常只能检测到细胞数量相对较多的微生物物种的存在,带谱的亮度在一定程度上可以反映出其所代表的物种的细胞数量,可以认为带谱中较亮的条带代表了环境中的优势菌群,说明这几种细菌在整个小麦生长期,虽然有数量上的变化,但一直对土壤肥力的稳定性乃至小麦的生长发育起着重要作用.在小麦不同的生育时期都有不同的细菌出现,整体表现为随着小麦的生长多样性逐渐增加,而到收获后又有所回落,与纯培养研究及其他一些作物的研究结果相一致[5].这可能与小麦不同时期的对营养的吸收,根系物质的分泌及季节变化有关.此外,研究还发现不同层次的土壤在同一时期多样性变化不明显,在不同时期变化差异较大.表层土壤各处理间菌群数量变化较大,而深层土壤处理间菌群数量变化较小.这种结果与传统的纯培养方法研究的结果有一定差异,传统方法显示随着土壤的深度加大,微生图4不同土样16S rDNA条带图谱的聚类分析F i g.4C l u ster analysis of16S rDNA banding p rofiles forco mmun ities fro m d ifferen t soil sa mp les物数量逐步递减[6].可能是由于在纯培养过程中一些细菌丢失,造成了对其多样性估计偏低所致,另外由于所研究土壤的深度不同,也会造成对土壤细菌群落数量估计的差异.可见,从分子水平对微生物的多样性及种群动态变化进行研究,能突破传统纯培养方法的局限性,更好的反映土壤微生物的真实存在状态.通过PCR-DGGE对小麦不同生育时期及不同深度土壤细菌多样性的分析,能够更真实的反映冬小麦-夏400河南农业大学学报第41卷玉米农作模式下对农田土壤细菌群落的影响,从而为该农作方式的改进提供理论指导,并根据群落种类的变化开发相应的新型农田微生物肥料.参考文献:[1]W ARD D M,W ELLER R,BATESON M M.16S r DNAsequences revea l nu m erous uncultured m icroorgan is m sin a comm un it y[J].N a t ure,1990(345):63-65. [2]EDR IC K D B,PEA COCK A,STEPHEN J R,e t a.lM easur i ng so il m icrob i a l community diversity using po-larli p i d fa tty aci d and denaturi ng g rad i ent g el electropho-res i s data[J].Journal o f M icrob i o log ica l M ethods,2000,41:235-248.[3]ZHOU J Z,M ARY A B,J AM ES M T.DNA recoveryfrom so ils of d i ve rse co m po siti on[J].Apple and Env-iron m entM icrobio l ogy,1996,62:316-3221[4]BASS AM B J,CAETANO-ANOLLES G,GRESS HOFF PM.Fast and sensiti ve silver sta i ni ng o fDNA i n po l yacry-la m i de ge l s[J].Anal Bioche m,1991(196):80-831[5]胡元森,吴坤,刘娜,等.黄瓜不同生育期根际微生物区系变化研究[J].中国农业科学,2004,37(10):1521-15261[6]贾志红,杨珍平,张永清,等.麦田土壤微生物三大种群数量的研究[J].麦类作物学报,2004(24):53-561(责任编辑:丁丽)。
本科生毕业论文(设计)题目: 基于Python的对土壤细菌群落结构的影响的16srRNA基因测序分析姓名: 熊艺学院: 资源与环境科学学院专业: 农业资源与环境班级: 资环132班学号: 13613229指导教师: 兰平职称: 研究员2017年5月16日南京农业大学教务处制目录摘要: (4)关键词: (4)Abstract: (4)引言 (5)1 材料与方法 (5)1.1 土壤样品介绍与处理方法 (5)1.2 Python语言以及生物信息相关Python包和软件的介绍 (6)1.2.1 Python在生物信息方面的应用 (6)1.3 Python开发环境搭建以及Biopython包的安装和使用 (6)1.3.1 安装python和相关的科学计算的包Python (6)1.3.2 搭建基于sublimetext3的python开发环境 (6)1.3.3 Biopython包的使用方法[9] (7)1.4 Qiime微生生物基因组分析工具的安装[10] (8)1.5 本研究使用的其它的Python包的简要介绍 (8)1.5.1 Numpy (8)1.5.2 Pandas (8)1.5.3 Scipy (8)1.5.4 Matplotlib (8)1.5.5 ETE Toolkit (8)1.5.6 NetworkX (8)1.5.7 Pygraghviz (8)1.6 进入Qiime工作流前测序原始数据的初步处理 (9)1.6.1 提取barcode序列 (9)1.6.2 reads拼接 (9)1.6.3 fasta文件拼接 (9)1.6.4 割库 (9)1.6.5 去除嵌合体序列 (10)1.7 OTU聚类和数据的分析和可视化 (11)1.7.1 OTU聚类 (11)1.7.2 OTU聚类和系统发育树的可视化 (11)1.8 多样性分析 (12)1.8.1 α-多样性分析 (12)1.8.2 β-多样性分析 (13)1.9 存在显著性差异物种分级聚类并可视化 (13)2 结果与分析 (13)2.1 土壤样品基本理化性质的分析 (13)2.2初步处理后的序列的质量控制结果 (14)2.3 OTU聚类和数据的分析和可视化结果 (14)2.4多样性分析结果 (14)3 讨论 (15)3.1 土壤理化性质的改变和土壤细菌群落结构变化的关系 (15)3.2 16s rRNA基因测序技术在土壤微生物生态研究中的局限性 (16)3.3 Python在16s rRNA基因测序数据分析中的优缺点 (16)致谢 (16)参考文献 (16)附录 (18)基于Python的对土壤细菌群落结构的影响的16srRNA基因测序分析农业资源与环境专业学生熊艺指导教师兰平摘要:施肥措施可以对土壤细菌群落结构以及其多样性产生影响,16s rRNA基因测序技术又是研究土壤样品中细菌群落组成结构的重要手段之一,而Python在分析生物序列数据方面具有软件资源丰富、语法简练的优点。
红树林土壤微生物的研究:过去、现在、未来蒋云霞;郑天凌;田蕴【期刊名称】《微生物学报》【年(卷),期】2006(46)5【摘要】红树林土壤生境的独特性决定了其中微生物的多样性及其资源的珍稀性,对于红树林土壤微生物的研究正在成为热点.然而由于传统研究方法等因素的限制,至今人们对红树林土壤微生物的系统了解仍较为有限.近年来,基于16S rRNA,18S rRNA基因的各种分子微生物学技术的迅速发展,红树林土壤微生物的研究亦面临着崭新的局面.文中主要从红树林土壤微生物物种的多样性、生理生化类型的多样性及其在治理污染环境、生物修复作用中的可能性、有效性等方面阐述了红树林土壤微生物的研究进展,并以更合理、有效地开发利用红树林土壤微生物资源为目标,展望了21世纪,以新理念、新技术、新方法进行红树林土壤微生物研究及资源开发的巨大前景.【总页数】4页(P848-851)【作者】蒋云霞;郑天凌;田蕴【作者单位】厦门大学,生命科学学院应用与环境微生物研究所,厦门,361005;厦门大学,生命科学学院应用与环境微生物研究所,厦门,361005;厦门大学,近海海洋环境科学国家重点实验室,厦门,361005;厦门大学,生命科学学院应用与环境微生物研究所,厦门,361005【正文语种】中文【中图分类】S7【相关文献】1.STS 研究的过去、现在与未来--爱丁堡大学科学技术与创新研究所 Robin Williams 教授访谈 [J], 胡明艳;简力;Robin Williams2.移动电子商务研究的过去、现在和未来——还有哪些领域需要继续研究? [J], Key Pousttchi;David Tilson;Kalle Lyytinen;Yvonne Hufenbach;盛小芳;唐红涛3.旅游消费者行为研究的过去、现在和未来——基于引证研究法的研究 [J], 张梦;郭养红;付晓蓉4.海峡论坛促进了海峡传播研究——两岸学者在第十届海峡论坛举办之际笔谈海峡传播研究的过去、现在与未来 [J], 李展5.心血管疾病危险因素的研究:过去、现在和未来 [J], 赵冬因版权原因,仅展示原文概要,查看原文内容请购买。
随着人们对环境问题的日益关注,从20世纪70年代开始,微生物生态学得到了迅速的发展[1.2]。
传统的微生物生态学技术包括显微形态观察,选择性培养基计数,纯种分离和生理生化鉴定[3]等。
但随着研究的深入,人们发现自然环境中存在的大多数微生物不可培养,传统的方法不能够满足微生物生态研究的需要,使微生物生态研究一直较为落后。
近年逐步建立起来的16SrRNA/DNA实验技术则为微生物生态学的研究提供了新的研究方法,建立了对难以培养的微生物和不依赖于培养的微生物生态学的研究方法,使关于微生物多样性、微生物种群分析、重要基因的发现、以及遗传物质在微生物之间或微生物与植物之间的水平转移对生态系统的影响等方面都取得很大进展,开辟了微生物生态学研究新的领域。
1微生物多样性研究微生物多样性的研究是微生物生态学研究的基本内容,由于微生物具有生存环境多样,生长繁殖快,适应性强,代谢途径多样以及生活方式多样的特点,使得微生物分布广泛,变异快,种类多,通过对微生物多样性的研究,可以探知蕴藏其中的大量基因资源。
因此对微生物多样性的研究,无论对于生态系统功能的完整理解还是对微生物资源的利用,都具有极其重要的意义[4]。
但是,传统的分析方法是通过分离、培养和鉴定,需要一个非常复杂的形态特征鉴定和生理生化试验,这种方法试验周期长,工作量大,更重要的尽管是最复杂的组合,也难于将样品中的微生物全部分离,而且不能对分离物进行准确的鉴定,不能反映分离物之间的系统发育关系[5]。
用分子生态学的方法可以直接从核酸水平对样品进行研究,通过提取样品中不同微生物的总DNA,然后进行分析,避免了样品中微生物的丢失,样品序列的多样性和不同序列的丰度在一定程度上反映了原始样品中微生物种群的多样性和丰度。
从而可以保证实验能更真实地体现研究对象的结构组成,因此分子生态学技术和研究策略在揭示自然环境中微生物多样性的真实水平及其物种组成上显示出了极大的优越性。
通过以土壤细菌16SrRNA基因的一段26碱基变异区为基因标签,连接成基因标签串测序,用标签类基于16SrRNA/DNA分析的土壤微生物生态学效应黄进勇,周伟(郑州大学生物工程系,河南郑州450001)摘要:基于16SrRNA/DNA分析的实验技术为微生物生态学的研究提供了新的研究方法,从而在微生物多样性、微生物种群分析、重要基因的发现以及遗传物质在微生物之间或微生物与植物之间的水平转移对生态系统的影响等方面都取得了很好的研究进展。
关键词:分子生态;微生物生态;16SrRNA/DNA中图分类号:Q938文献标识码:AEcologicalEffectonSoilMicrobesontheBasisof16SrRNA/DNAAnalysesHuangJinyong,ZhouWei(Bioengineeringdepartment,Zhengzhouuniversity,Zhengzhou450001)Abstract:The16SrRNA/DNAtechniquehasofferedthenewapproachtoresearchonmicrobialecology.Onthebasisofthemethod,someconsiderableprogresshasbeenmadeinsomeaspectssuchasmicrobesdiversity,microbespopulationanalyses,discoveryofsomeimportmentgenes,andeffectonecosystemofhorizontalmovementofhereditarypropertybetweenthemicrobesorbetweenmicrobesandplants.Keywords:Molecularecology,Microbialecology,16SribosomeRNA/DNA基金项目:河南省自然科学基金资助项目(0511530400)。
第一作者简介:黄进勇,男,1969年出生,河南周口人,博士学历,副教授,主要从事生态学、生物多样性研究。
E-mail:jinyhuang@zzu.edu.cn。
收稿日期:2005-12-14。
型、频率和多样性指数在分子水平描述土壤细菌遗传多样性,分析了土壤中细菌遗传多样性与植被类型之间相关性,发现土壤细菌遗传多样性与土壤有机质、全氮含量等理化性质高度相关,不同的植被土壤理化性质差异决定细菌遗传多样性分化[6,7]。
通过直接从土壤中抽提总DNA,并对DNA中V-3可变区进行PCR扩增,然后进行变性梯度凝胶电泳等对农田土壤微生物种类分布进行研究,陈灏等发现不同的农田土壤间细菌差异相当显著,同时他们通过对分离带的测序,进行了部分土壤微生物的系统分类,为农田土壤生态和高效氮肥的研究提供了新的实验依据[8]。
有学者用DGGE法分析了镉胁迫下稻田土壤微生物的多样性,发现土壤镉有效性的变化强烈影响土壤微生物基因多样性的变化,随着土壤中镉浓度的增加,对土壤中微生物基因多样性的影响也逐渐增强[9]。
Torsrik等研究了挪威等地的森林土壤样品,通过提取样品中的总DNA,然后用高压下将其剪接成分子量平均为420kD的DNA片段,测定热复性过程,得到Cot1/2的值。
结果显示,该土壤样品基因多样性是一般分离培养情况下得到的多样性的200多倍,显示出分子生态学技术在微生物多样性研究中的优越性[10]。
Salles等用设计的伯克氏菌属(Burkolderik)特异性引物进行DGGE检测来分析两块草地中该属群落的多样性,揭示了主体与根际土壤样品中生物多样性的不同[11]。
2微生物的分类和鉴定微生物的分类和鉴定与动植物相比,存在着较大的弊端,一方面它形态较小,需要借助显微镜观察,另一方面自然界中还存在着大量的不能培养的微生物,这就给微生物的分类与鉴定带来极大的麻烦,而分子生态学的应用则正好为微生物分类提供了一个解决的途径。
通过对提取样品中特定的核酸片段,然后进行序列分析或比对,从而对未知微生物进行鉴定,并进一步确定其分类地位。
采用细菌16SrDNA通用引物和PCR扩增等方法,许飞等构建了南海南沙海区沉积物16SrDNA文库,并通过RFLP酶切分型对所获得的70个克隆进行测序,通过构建序列同源性矩阵和系统发育树图,发现该海域沉积物中变形细菌是明显的优势类群,α-Pro-teobacteria是深海沉积物中所特有的类群,是深海沉积微生物中的标志类群。
同时,作者从南沙海区获得的大多数16SrDNA序列,通过序列比对发现,所得的序列与发表在数据库中的16SrDNA序列表现出较大的差异,说明这些细菌在属种水平上完全不同,在这些未知种群中,蕴藏着目前还无法估量的资源,从而为开发利用这一特殊生态环境的微生物资源奠定基础[12]。
Nielsen用DGGE法研究变质的生物磷去除反应器中微生物的多样性,通过序列分析切下的电泳带,发现了一类与已知种类无密切关系的γ-变形菌[13]。
中国学者徐平等用包含16SrRNA基因v-2高变区在内的120bp长核苷酸序列,对300多株已知该区段序列的链霉菌进行分析,得到较为完整的系统进化树,确定了他们的分类地位[14]。
此外,通过对化学生物絮凝池活性污泥中优势菌种16SrDNA序列的分析,研究者发现这些优势菌种大多属于好氧菌群,而且基本上可以分为Acinetobacter属和Arcobacter属2大支[15]。
有人用DGGE法和纯培养法,对青海柯柯盐湖、茶卡盐湖底泥及周边土壤样品的细菌多样性进行了研究。
结果显示,两个盐湖存在大量的未知细菌,分离到的纯培养仅占实有细菌的小部分。
对12个菌株的16SrRNA基因克隆测序结果显示,其中一个属于Halomonadaceae科Halomonas属,其余菌株均属于Bacillaceae科,分属于Halobacillus、Sal-ibacillus、Amphibacillus、Virgibacillus、Gracilibacillus、Bacillus等6个属,从而确定青海盐湖确实存在大量的未知微生物[16]。
3微生物在环境检测与修复中的作用关于污染物对环境影响的生态效应,需要一个可被广泛采用并较准确的评价标准。
由于微生物对环境的适应性强,在各种环境中普遍存在,能对环境的微小变化做出迅速反应,所以可用分子生态学的方法在分子水平对微生物区系变化进行分析,从而更准确更科学地对环境污染状况进行评估。
此外,微生物在对环境的适应方面具有独特的优越性性,它能够迅速适应环境的变化并对不利的环境条件进行修复,使生态维持平衡。
因此,用分子方法研究不同污染条件下的微生物,可以发现对特定污染物具有修复作用的微生物或基因资源,进而通过对特殊菌株的选育,或对这些基因资源进行开发利用,使其在污染物的修复中发挥独特优势,为环境修复工程提供良好的工具。
通过提取南极阿德雷岛沉积物柱状样各层次的总DNA,利用PCR-RFLP方法,对沉积物中的细菌多样性及分布进行微生物生态系统已经受到了人类活了研究,发现大量与降解有机化合物相关的细菌,表明阿德雷岛受到人类活动的影响,但通过实验仅发现少量与重金属有关的菌株存在,说明该地区的重金属污染较轻,还没有严重影响微生物群落组成[17]。
有学者通过16SrDNAV3-PCR/TGGE指纹图谱技术对一个焦化工业废水处理系统两个功能不同的曝气池活性污泥的细菌种群结构进行了动态分析。
结合条带回收、克隆、序列测定等方法对TGGE条带的序列多态性和主要功能曝气池细菌种群多样性进行了探讨,以全面了解活性污泥处理系统细菌群落结构和功能动态变化关系,为实现对活性污泥优化操作奠定方法和理论基础[18]。
CoskunerG和CurtisTP于2002年用FISH技术原位鉴定了活性污泥中硝化细菌群落的组成和分布,为理解污水处理中氮素循环提供了重要的信息[19]。
Duarte通过PCR-DGGE研究取自含硫油浓度梯度污染的土壤和用含二苯噻吩石油处理的土壤小生态系的样品,结果显示,随着土壤中油含量的增加,分子群落轮廓中所检测带的数目减少,且含DBT石油处理后的分子群落随时间逐步改变,而未处理的多半是稳定的,这为检测石油对土壤的污染状况提供了新的思路[20]。
中国学者姚健等通过RFLP法对农用化学品污染的土壤微生物群落进行了分析,发现农用化学品的使用会改变土壤微生物群落DNA序列组成,并对微生物DNA序列多样性影响各不相同:在化肥污染条件下与无污染的土壤相比,土壤微生物生物量大大增加,而且化肥使微生物富集而一些物种丧失,而农药使微生物多样性增加[21]。
杨元根等通过对某一重金属污染土壤的微生物DNA进行DGGE电泳,其电泳谱带显示,与DNA标志物相比,土壤样品微生物的DNA片段谱图明显不同,反映了它们之间DNA基因型的显著差异;而不同土壤样品间DNA反映出来的DGGE谱带却非常一致,说明其微生物在DNA上没有显著的改变,反映了该地区土壤中重金属的积累并没有导致土壤微生物在基因上的损伤[22]。