生工在线订购引物单
- 格式:xls
- 大小:28.00 KB
- 文档页数:3

引物的应用常识引物的原理引物是短的寡核苷酸片段,充当DNA复制的起点。
因为几乎所有DNA聚合酶都不能从头合成,所以它们需要一个3’-羟基作为DNA合成的起始点。
这个3’-羟基由相配的引物提供。
在体内,由于DNA聚合酶的忠实性,不能从头合成DNA,因此只能由RNA聚合酶(称为引物酶)生成,采用RNA引物来延伸,在延伸过程中,RNA引物降解并由DNA取代。
在体外PCR反应中所用到的DNA引物,是根据不同的要求及模板序列设计,然后用化学法人工合成的,与模板形成双链后在DNA聚合酶的作用下就可以继续链的延伸;对于大多数PCR反应,决定整个反应成功与否的最重要因素是引物的序列和质量。
1. 不同实验要求的引物选择在开始设计引物之前,必须弄清以下几点:(1)明确PCR的目的(例如克隆、SNP检测、定量检测等)(2)确定样品材料(基因组DNA、RNA、微小RNA)(3)确定PCR的类型(普通的、定量PCR、RT-PCR、长片段PCR),在查找序列的时候还需要考虑可能存在的问题(如假基因等)2.引物设计的重要因素有一些不同的软件工具可用于引物设计和引物分析。
引物设计的软件如Oligo 6.22 ,Premier 5.0,Primer Express 3。
引物分析常用Primer 5,Oligo 6.22,Primer-Blast。
目前生工生物给客户提供的引物设计服务引物用的是在线软件Primer 3 plus,•引物长度和专一性常见的引物长度为18-30个碱基。
短的引物(≤15碱基)能非常高效地结合, 但是它们的专一性不够。
较长的引物能提高专一性,然而退火效率低,从而导致PCR产量低下。
同时应避免编码单一序列和重复序列的引物。
•平衡GC含量,避免GC-和AT-富集区域引物的GC含量应介于40%~60%之间。
应避免聚-(dC)-或聚(dG)-区域,因为它们会降低退火反应的专一性。
聚-(dA)-和聚(dT)-也应避免,因为这样会形成不稳定的引物-模板复合物,从而降低扩增效率。
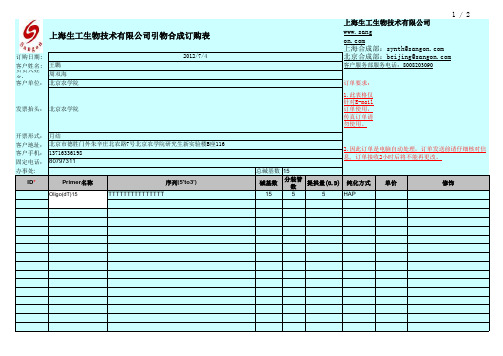
订购日期:
客户姓名:负责人姓名:客户单位:订单要求:
发票抬头: 1.此表格仅针对E-mail订单使用,传真订单请勿使用。
开票形式:客户地址:客户手机:137********固定电话:办事处:
总碱基数ID *
Primer 名称
序列(5'to3')
碱基数分装管
数
提拱量(O.D)纯化方式
单价修饰
Oligo(dT)15
TTTTTTTTTTTTTTT
1555HAP
15
北京农学院北京农学院北京市德胜门外朱辛庄北农路7号北京农学院研究生新实验楼B座116 2.因此订单是电脑自动处理,订单发送前请仔细核对信息, 订单接收2小时后将不能再更改。
80797311月结
周双海
上海生工生物技术有限公司引物合成订购表
上海生工生物技术有限公司上海合成部:synth@ 2012-7-4北京合成部:beijing@
王鵬客户服务部服务电话:8008203090。
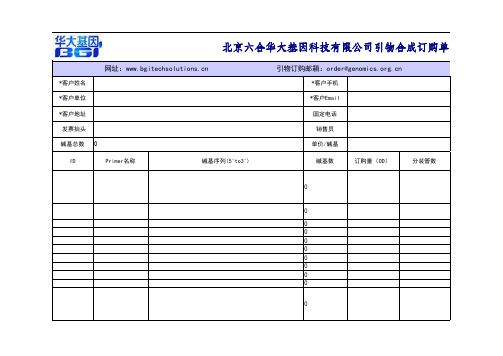


引物溶解、分装需要注意的首先拿到引物是要先快速离心一下,3000转1分钟,然后小心开盖,加水溶解,至于加什么水,有几种不同观点,加好水后用盖上盖子来回混匀5-10分钟,也可用漩涡振荡器,但不要太久,我震荡了好久,结果杯具了。
最后储存于-20℃中,避免反复冻融。
观点1:引物在碱性条件下保存时间较长,双蒸水偏酸性,DNA容易降解,TE Buffer pH在7.5-8左右,弱碱性,所以我们建议用1×TE Buffer 溶解引物,并保存于-20度。
观点2:TE是弱碱性,适合于引物的保存,引物若在酸性下比较不稳定。
用ddH2O 的话,更适合于后续的PCR操作。
观点3:注意工作液千万别用TE稀释,要用DEPC水,或去离子水稀释.因为TE可以鏊合镁离子,镁离子对Taq酶活性发挥至关重要,影响PCR反应.。
小结:买试剂公司做好的无DNA/RNA酶的水溶解最好,但是那个水的PH是多少我还不知道,下周一会送来,我测一下哈~另一个解决办法是:TE水溶解成100uM储存液,利于保存,用时每次做100ul或者50ul的稀释,用灭菌的ddH2O。
TE Buffer的配制方法组成浓度:10mM Tris-HCl1mM EDTA PH=8.0配制量:500ml配制方法:量取下列溶液于500ml烧杯中1M Tris-HCl Buffer PH=8.0 5ml0.5M EDTA PH=8.0 1ml向烧杯中加入约400mldd H2O均匀混合;将溶液定容到500ml后,高温高压灭菌;室温保存.。
相关技术资料来自上海生工网站:1.如何测定引物的OD值?用紫外分光光度计在260nm波长测定溶液的吸光度来定量,请注意紫外分光光度计的使用,测定时溶液的吸光度最好稀释到0.2-0.8之间(吸光度太高或太低会有较大的误差)。
DNA干粉用一定体积的水充分振荡溶解以后,取部分溶液稀释到1ml并在1ml标准比色皿中测定其吸光度,即为所测体积的OD值,进而可以计算出母液的OD值。
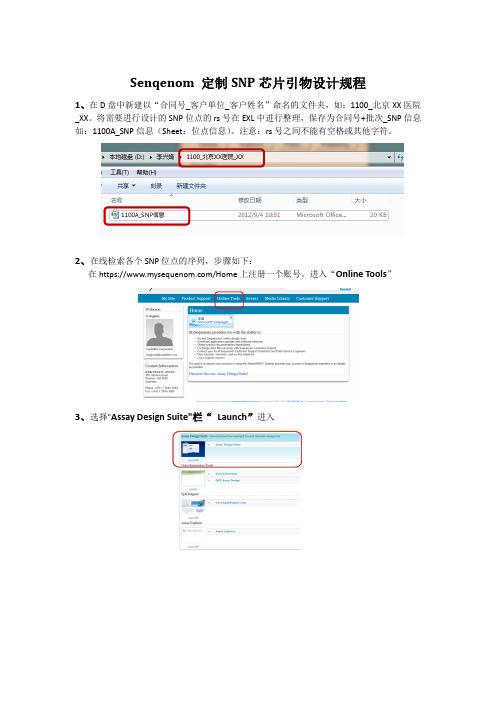
Senqenom 定制SNP芯片引物设计规程1、在D盘中新建以“合同号_客户单位_客户姓名”命名的文件夹,如:1100_北京XX医院_XX。
将需要进行设计的SNP位点的rs号在EXL中进行整理,保存为合同号+批次_SNP信息如:1100A_SNP信息(Sheet:位点信息)。
注意:rs号之间不能有空格或其他字符。
2、在线检索各个SNP位点的序列,步骤如下:在https:///Home上注册一个账号。
进入“Online Tools”3、选择"Assay Design Suite"栏“Launch”进入4、出现“Genotyping Design ”设计界面,如下图Design Name:合同号+批次或客户姓名拼音Vesion:设计版本(1,2,3...)rs or FASTA:在此输入SNP位点的rs号。
Orgnism:物种Database:数据库版本Chemistry :iPLEXMultiplex Level: 36 (每个WELL反应重数)5、点击“Edite Text Input”,将整理好的rs号粘贴至对话框中,点击“Save”,如图:6、根据检测需求单中样品的信息,更改“物种,数据库版本,反应重数”7、以上参数修改完成后,在第“4 Design Assays”项中右击选择“Run up to the previous step”进行前3项的在线运行。
运行结束后如下图:8、点击“Export All”出现如下窗口,选择“Export All Results”,保存在步骤1中建立的该项目文件夹中。
命名格式为:合同号+批次_设计版本(V1/V2/V3)。
9、右击该压缩文件,解压缩后如图,含有三个文件夹:Pretend,Proxsnp,Sequence Retrieval。
10、"Pretend"文件下有2个“1100A_V1”文本,一个为本次在线检索的信息,另一个“1100A_V1”文本为本次检索的“SNP_ID Sequence”信息,如图。
引物的应用常识引物的原理引物是短的寡核苷酸片段,充当DNA复制的起点。
因为几乎所有DNA聚合酶都不能从头合成,所以它们需要一个3’-羟基作为DNA合成的起始点。
这个3’-羟基由相配的引物提供。
在体内,由于DNA聚合酶的忠实性,不能从头合成DNA,因此只能由RNA聚合酶(称为引物酶)生成,采用RNA引物来延伸,在延伸过程中,RNA引物降解并由DNA取代。
在体外PCR反应中所用到的DNA引物,是根据不同的要求及模板序列设计,然后用化学法人工合成的,与模板形成双链后在DNA聚合酶的作用下就可以继续链的延伸;对于大多数PCR反应,决定整个反应成功与否的最重要因素是引物的序列和质量。
1. 不同实验要求的引物选择在开始设计引物之前,必须弄清以下几点:(1)明确PCR的目的(例如克隆、SNP检测、定量检测等)(2)确定样品材料(基因组DNA、RNA、微小RNA)(3)确定PCR的类型(普通的、定量PCR、RT-PCR、长片段PCR),在查找序列的时候还需要考虑可能存在的问题(如假基因等)2.引物设计的重要因素有一些不同的软件工具可用于引物设计和引物分析。
引物设计的软件如Oligo 6.22 ,Premier 5.0,Primer Express 3。
引物分析常用Primer 5,Oligo 6.22,Primer-Blast。
目前生工生物给客户提供的引物设计服务引物用的是在线软件Primer 3 plus,•引物长度和专一性常见的引物长度为18-30个碱基。
短的引物(≤15碱基)能非常高效地结合, 但是它们的专一性不够。
较长的引物能提高专一性,然而退火效率低,从而导致PCR产量低下。
同时应避免编码单一序列和重复序列的引物。
•平衡GC含量,避免GC-和AT-富集区域引物的GC含量应介于40%~60%之间。
应避免聚-(dC)-或聚(dG)-区域,因为它们会降低退火反应的专一性。
聚-(dA)-和聚(dT)-也应避免,因为这样会形成不稳定的引物-模板复合物,从而降低扩增效率。
引物纯化方式选择指南2012-2-16 10:24:14内容导读一、DNA合成的方法和原理二、引物纯化的方法原理及其效果三、纯化方法与应用指南四、常见问题的原因分析及相应的对策一、DNA合成的方法和原理目前引物合成主要采用固相亚磷酰胺三酯法进行。
基于该方法的DNA合成仪有多种,由ABI/PE公司生产的高通量DNA自动合成仪得到了广泛的应用。
各合成仪进行引物合成的原理基本相同,主要区别在于合成产率的高低、试剂消耗量和单个循环用时等。
生工公司采用的合成仪主要机型为全新的ABI3900高通量合成仪。
固相亚磷酰胺三酯法合成DNA片段,具有高效、快速偶联以及起始反应物比较稳定的特点。
该方法是在固相载体上完成DNA链的合成的,DNA化学合成不同于酶促的DNA合成过程从5’ →3’方向延伸,而是由3’端开始,相邻的核苷酸通过3’→ 5’磷酸二酯键连接。
具体的反应步骤如图一。
1、脱保护基(Deblocking)用三氯乙酸(Trichloroacetic Acid,TCA) 脱去连结在CPG (Controlled Pore Glass) 上的核苷酸的保护基团DMT (二甲氧基三苯甲基),获得游离的5'-羟基端,以供下一步缩合反应。
2、活化(Activation)将亚磷酰胺保护的核苷酸单体与四氮唑活化剂混合并进入合成柱,形成亚磷酰胺四唑活性中间体(其3'-端已被活化,但5'-端仍受DMT保护),此中间体将与GPG上的已脱保护基的核苷酸发生缩合反应。
3、连接(Coupling)亚磷酰胺四唑活性中间体遇到CPG上已脱保护基的核苷酸时,将与其5'-羟基发生亲合反应,缩合并脱去四唑,此时合成的寡核苷酸链向前延长一个碱基。
4、封闭(Capping)缩合反应后,为了防止连在CPG上的未参与反应的5'-羟基在随后的循环反应中被延伸,常通过乙酰化来封闭此端羟基,一般乙酰化试剂是用乙酸酐和N-甲基咪唑等混合形成的。