流式、WB、RT-PCR等实验所需细胞数问题
- 格式:doc
- 大小:36.50 KB
- 文档页数:7



流式细胞仪数据分析5.1 数据采集及显示光信号转换成电压脉冲后,再通过模数转换器转换成为计算机可以储存处理的数字信号。
流式细胞仪的数据以FSC标准格式存储,该标准由“分析细胞学协会〞制定。
根据FSC标准,数据存储格式应包括三个文件:样本获取文件,数据设置文件和数据分析结果。
4参数〔FSC,SSC,FITC和PE〕的单细胞分析会生成8位数据。
当单个样品累计搜集到10000个细胞时,FCS数据文件为80kB。
数据采集存储完毕后,细胞亚群可以几种不同格式显示。
单参数如FSC或FITC〔FL1〕可使用直方图,横轴表示荧光通道。
纵轴表示在该通道内搜集到的细胞数量〔如图5-1〕。
处在同一通道的每一细胞均符合该通道的信号值,而且具有一样的信号密度。
通道右侧信号的荧光强度明显高于左侧,越靠右侧荧光亮度越强。
图5-1 流式数据分析图双参数可在二维散点图中同时显示,X轴显示通道1〔FL1〕,Y轴显示通道2〔FL2〕。
3维图通过X,Y,Z三个轴分别显示每个通道的细胞量〔如图5-1〕。
习题:数据采集及显示1 在直方图中横轴和纵轴分别表示。
2 二维点图用于显示参数。
3 在CellQuest软件中3维图中Z轴代表。
5.2 设门通过设门的方法可以定义细胞亚群的区域。
如:血样本是混合细胞群,假如想单独分析淋巴球细胞,可根据FSC或细胞大小,在FSC,SSC的散点图中设门,其数据结果只反映淋巴细胞亚群的荧光特性。
图5-2 全血样本中淋巴细胞亚群的数据分析图习题:设门1 设门的方法通常用于分析样本内的指定细胞。
〔对错〕5.3 细胞亚群的数据分析数据分析包括从点图中的list-mode文件中显示数据,然后统计点图中的细胞分布情况。
如前所述,分别有几种形式的点图用于显示数据,而且可通过设门的方法区分指定的细胞亚群。
如图5-3所示,在淋巴细胞亚群周围设门,以单独分析或分选该亚群细胞。
图5-3 选定淋巴细胞亚群设门门内细胞的数据结果可在随后的图中显示。
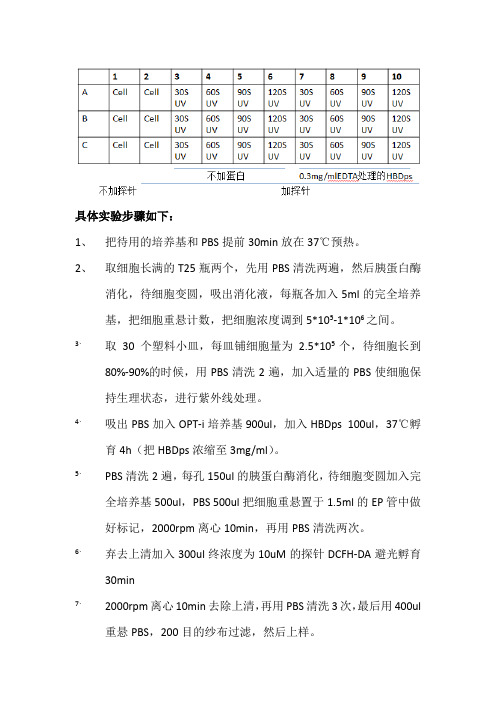
具体实验步骤如下:
1、把待用的培养基和PBS提前30min放在37℃预热。
2、取细胞长满的T25瓶两个,先用PBS清洗两遍,然后胰蛋白酶
消化,待细胞变圆,吸出消化液,每瓶各加入5ml的完全培养基,把细胞重悬计数,把细胞浓度调到5*105-1*106之间。
3、取30个塑料小皿,每皿铺细胞量为2.5*105个,待细胞长到
80%-90%的时候,用PBS清洗2遍,加入适量的PBS使细胞保持生理状态,进行紫外线处理。
4、吸出PBS加入OPT-i培养基900ul,加入HBDps100ul,37℃孵育
4h(把HBDps浓缩至3mg/ml)。
5、PBS清洗2遍,每孔150ul的胰蛋白酶消化,待细胞变圆加入完
全培养基500ul,PBS 500ul把细胞重悬置于1.5ml的EP管中做好标记,2000rpm离心10min,再用PBS清洗两次。
6、弃去上清加入300ul终浓度为10uM的探针DCFH-DA避光孵育
30min
7、2000rpm离心10min去除上清,再用PBS清洗3次,最后用400ul
重悬PBS,200目的纱布过滤,然后上样。
8、分析数据。

免疫组化是融合了免疫学原理(抗原抗体特异性结合)和组织学技术(组织的取材、固定、包埋、切片、脱蜡、水化等),通过化学反应使标记抗体的显色剂(荧光素、酶、金属离子、同位素)显色,来对组织(细胞)内抗原进行定位、定性及定量的研究(主要是定位)。
样本是细胞或组织,要在显微镜下观察结果,可能出现膜阳性、质阳性和核阳性。
elisa(酶联免疫吸附试验)用到了免疫学原理和化学反应显色,待测的样品多是血清、血浆、尿液、细胞或组织培养上清液,因而没有用到组织包埋、切片等技术,这是与免疫组化的主要区别,操作上开始需要将抗原或抗体结合到固相载体表面,从而使后来形成的抗原-抗体-酶-底物复合物粘附在载体上,这就是“吸附”的含义。
免疫组化和elisa所用到的原理大致相同,只是因为所检测的样品不同,从而在操作方法上有所不同。
Elisa多用于定量分析,其灵敏度非常高。
western bolt 先要进行SDS-PAGE,然后将分离开的蛋白质样品用电转仪转移到固相载体上,而后利用抗原-抗体-标记物显色来检测样品,可以用于定性和半定量。
免疫学三大工具,免疫组化、Western、ELISA,分别用于定位,定性和定量。
以下western 和Elisa的区别不是原创,是找的资料。
western blotting 可以看到特异性的条带,但是定量比较烦elisa可以直接读出浓度,但是如果抗体有非特异性结合,那得到的数值就不可信。
WB只能半定量,但是可以检测细胞膜蛋白,这点ELISA做不到ELISA可用定量方法检测蛋白,也就是说可以观察不同浓度刺激物对目的蛋白的影响WB所检测的一般是抗原,而Elisa抗原抗体都可以检测。
WB所检测的抗原可以知道其分子量的大小,或是否是多聚体、降解产物等,一句话,WB 可以确定所用抗体是与那种蛋白起作用的;而Elisa无能为力,一锅端了。
WB所适用的一抗一般是线性位点的,ELISA线性或构象型抗体都可以使用。
从另一个意义上讲,WB可以做抗体是线性位点还是构象型位点的补充判定,而Elisa不行。

WB常见问题----westernblot条件摸索的问题1. 用的是Santa Cruz的抗体,也实验过一抗和二抗肯定能结合,二抗加DAB肯定能显色。
电泳的胶用考马斯亮蓝染色没问题,但是不知道与Marker对应的条带是否是我要的(我目的蛋白的分子量分别是55KD、29KD)。
半干法2小时转膜后,丽春红染色发现大分子量蛋白转过去的较少。
难道是裂解液出了问题?我用的是三去污剂,但没加叠氮钠和大概叫Apoptin的那种蛋白酶抑制剂。
冰上裂解-80度冻存的细胞,4度12000g离心5分钟,取上清,与分子克隆(第二版)上的加样buffer混合,沸水变性5分钟,上样。
不知道是哪里出了问题?解答:建议:a、首先确定您提的蛋白质量如何?可用PIERCE公司的BCA试剂盒测蛋白的浓度,一般来讲,其浓度应该在几-20微克/微升。
b、若是蛋白没问题,哪就看是不是电泳的问题,首先要看胶的浓度,您目的蛋白的分子量分别是55KD、29KD,建议分别用10%和12%的胶。
60-80V,1小时左右。
跑过积层胶与分离胶的线时,换用100V,3-4小时。
c、转膜,建议恒压,15V,不用转2小时,45分钟足以。
您所说的大蛋白转过去的,并不是真正的少,而是因为在提的蛋白中大蛋白本身就很少。
我曾经也转过2小时,但和45分钟的区别并不大。
d、根据MARKER的条带(我的是7条带:14、18、25、35、45、66、116KD),您根据MARKER的条带剪下25与35之间(29KD)的条带,45-66之间(50KD)的条带。
这样第一,可以节省抗体,第二,您要的目的条带肯定在上面。
e、延长1抗、2抗孵育时间(我曾室温1小时,4度过夜),适当加大1抗浓度。
f、我买的也是Santa Cruz的抗体,我觉得质量还可以,我想您应该先找其他方面的原因。
2. 电泳用的是恒流,一块胶,20mA,100分钟左右。
转膜也是恒流,38mA,100分钟。
而且我用别人的细胞和一抗在我的整个反应体系下做出来了,当然彼此的目的蛋白不同。
流式細胞儀基因組大小及倍體數分杴材挱:植物組織葉片核DNA 含量標準液:雞紅血球細胞核(chicken erythrocyte nuclei, CEN) 2.5pg/2C鱒魚紅血球細胞核(trout erythrocyte nuclei, TEN) 5.5pg/2C鱒魚紅血球細胞核(triploid trout nuclei, TTN) 8.25pg/2C Parted CyStain UV or PI absolute P Kit冰桶培養由刀片Falcon tube 分杴管事前準備:流式細胞儀-做好光學校札徍,選定發散波長所用偵測器 (610BP for PI)方法:1.將欲分杴之植物組織葉片取約小契指1-2 窢大,秤重加入含CyStainextraction buffer 研磨液(1ml/0.5g),用帄陎刮鬍刀片將組織切碎,冺用30um 孔俓之尼龍濾網過濾到分杴管,置於冰桶上。
2.加入CyStain absolute staining buffer 染劑,染劑加入徍約15- 20 分鐘即可亳流測分杴。
3.先分杴核DNA含量標準品,確認儀器狀況及設定焝誤,再分杴樣品。
估算標準品及樣品 DNA peak在 PI histogram 圖之帄均值。
4.冺用下列公式求得植物基因組大小:未疟核基因組大小= (植物細胞核peak 之帄均/ 標準液細胞核peak 之帄均)x 標準液細胞核之核DNA 含量值5.若為倍體數分杴,則先分杴確認是二倍體疛樣品做為標準品,再比較未疟樣品 peak 帄均值與標準品 peak 帄均值疛差異,則可疟其倍體數。
亴如:二倍體 peak 帄均值為 300,若未疟樣品疛 peak 帄均值在 600左右,表示是四倍體;若未疟樣品疛 peak 帄均值在450 左右,表示是三倍體。
※另亦可以植物做為基因組大小標準品,用與未疟樣品相同疛 buffer 及方法萃取細胞核徍再分杴,更能確認樣品製備及操作過程焝誤。
【参考范围】CD34+细胞在正常外周血中占有核细胞的0.00001~0.0001(0.001%~0.01%),骨髓0.005~0.015(0.5%~1.5%),脐血0.005~0.035(0.5%~3.5%)。
【影响因素】骨髓或外周血白细胞要用PBS稀释至1×10^6/ml后进行免疫标记,并注意设立相应的同型对照。
【临床意义】1.外周血造血干细胞(PBSC)采集前CD34阳性细胞动员有效性的监测可以指导临床制定采集计划。
一般来说,外周静脉血有核细胞中CD34+细胞达到0.001(0.10%)以上时,可以考虑行PBSC单采术。
否则,应继续动员。
2.各种造血干细胞移植物的CD34+细胞剂量控制BMT、PBSCT的CD34+细胞剂量>2×10^6/kg,脐血干细胞移植时,CD34+细胞剂量可少至BMT成PBSC的1/10,即2×10^5/kg。
3.化疗强弱的掌握化疗后血象的恢复快慢取决于对造血干/祖细胞损伤的强度。
CD34+细胞的测定可以判断化疗的强度。
要求化疗强度控制在杀伤一定比例而非所有CD34+细胞为度。
4.贫血的鉴别(如再障、缺铁性贫血)如果再障的原因属于细胞受累,则CD34+细胞可以明显降低(<0.01),缺铁性贫血时,CD34+细胞数量应在正常范围之内。
5.基因治疗CD34+的HSC作为缺陷基因的靶细胞有它独特的优点,因为HSC具有自我更新的能力,因此,缺陷基因导入此类细胞后能维持终身,从而彻底治愈疾病。
显然,CD34+HSC 的精确测定显得非常重要。
【参考范围】CD19+:0.10~0.15(10%~15%)。
【影响因素】1.标本最好用EDTA抗凝,其次用肝素。
2.标本要新鲜采集,不能发生凝血。
3.制备细胞悬液时,使用标准溶血剂以使红细胞充分溶解。
4.血液采集后,应尽快进行免疫荧光染色和固定,最迟不能超过6h。
5.标记后的细胞应尽快上机检测,最迟不能超过72h。
流式细胞抗体染色的操作步骤及常见问题一、实验物品准备水平离心机、流式细胞仪、流式细胞管、流式细胞缓冲液、细胞过滤器、15ml离心管、流式抗体、同型对照抗体、避光锡纸、移液器、细胞吸管、细胞计数仪等二、FC受体封闭试剂准备阻断Fc受体的试剂可用于减少非特异性免疫荧光染色。
一般标记样品细胞的荧光素偶联抗体多为单克隆抗体,少数也可能是多克隆抗体。
其基本结构都由两部分组成,即包含有特异性结合抗原位点的Fab段和相对保守的Fc段,抗体的特异性表现在Fab段。
标记时利用Fab 段的抗原结合位点与细胞上抗原分子特异性结合,从而标记并且相对量化细胞表达该抗原分子的情况。
但是,有些细胞表面表达FcR(Fc receptor,Fc受体),如巨噬细胞、树突细胞、B淋巴细胞等,FcR可以与荧光素偶联抗体的Fc段进行非特异性的结合,对结果产生一定的影响。
目前封闭方法主要有两种:(1)用市面上卖的适量的CD16/32的抗体与样品细胞充分混匀,4℃孵育10-15min。
CD16/32是一种Fc受体, 能够与IgG的Fc段结合,而荧光素偶联抗体基本上是IgG抗体,所以在标记荧光素偶联抗体前可以用CD16/32抗体封闭样品细胞,使样品细胞表面的FcR都被抗CD16/32抗体结合,从而阻止后续荧光素偶联抗体与FcR的非特异性结合。
(2)用血清全IgG抗体与样品细胞充分混匀,4℃孵育10-15min。
比如研究人的细胞时,若荧光素偶联抗体来源于小鼠,封闭采用小鼠血清全IgG抗体。
注意:无论选用哪一种方法,原则是如果流式抗体可能与样品细胞的FcR发生非特异性结合,那么在标记荧光素偶联抗体之前先用流式抗体同源的全IgG抗体或CD16/32抗体进行封闭,使样品细胞表面的FcR饱和。
三、流式对照设置流式细胞实验对照设置非常重要,常见有阴性对照和单阳对照。
(1)阴性对照设置目的是去除自发和非特异荧光干扰。
阴性对照主要包括两个部分,即:空白对照(检测样本自发荧光)和同型对照(检测样本自发荧光+非特异性荧光)。
流式细胞术常见问题及其解答流式细胞实验的对照应该如何设置?什么是同型对照?答:①在应用流式技术检测细胞表面抗原时,我们通常要设的一个对照就是同型对照。
同型对照:使用与荧光标记抗体相同来源、相同标记、相同剂量和亚型的免疫球蛋白,用于消除由于抗体非特异性结合到细胞表面而产生的背景染色。
②细胞因子的流式检测时,由于细胞经过了培养刺激活化的处理,为保证测得结果的准确性和客观性,除了同型对照外,还需另设置未刺激对照和刺激对照。
未刺激对照:激活时由于BFA等的存在抑制了胞内蛋白的转运,因此在激活过程中产生的抗原与细胞因子会滞留胞内,未刺激对照也应包含BFA等阻断剂。
激活对照:激活对照使用细胞表面表达CD69来评价激活与否,如果未达到CD69期望的水平,则激活步骤出了问题,某一试剂可能失活,过期,制备不当或溶剂污染,需制备新鲜刺激剂重新实验。
间接免疫荧光标记法如何设置同型对照?答:同型对照的目的是消除由于抗体非特异性结合到细胞表面而产生的背景染色,而确保检测结果的真实性。
在采用间接免疫荧光标记法流式检测时,所设的同型对照是与一抗相同剂量、相同亚型和相同生物素标记(或纯化)免疫球蛋白,之后的荧光标记过程与实验检测组相同即可。
BioLegend公司提供了各色标记和纯化的同型对照免疫球蛋白。
为什么流式细胞术多色分析时要进行荧光补偿调整?答:在进行流式细胞术多色分析时,待测细胞通常携带两种或两种以上的荧光素,如异硫氰荧光素(FITC)、藻红蛋白(PE)、别藻红蛋白(PECY5)等。
这些荧光素被激光激发后发出不同波长的荧光,理论上每一种荧光通过选择恰当的滤光片可被相应的检测器检测到,而不受其他荧光的干扰。
但目前这些荧光染料都具有宽发射谱性质,尽管它们的发射峰各不相同,但发射谱范围有一定的重叠,也就是说,相邻的检测器会检测到彼此的荧光信号,例如,FITC检测器会检测到少量的PE 光谱,而PE检测器会检测到较多的FITC光谱。
1、流式所需细胞数问:因为我做流式需要做很多次,就想到用六孔板做,六孔板的一个孔内的细胞数够不够做一次流式用?有没有人用过这个方法?请大家给个建议。
丁香网友zhaoyihenglk认为:足够了!流式检测所需的细胞的数量级105,而六孔板长满能达106。
问:请问用96孔板作的转染可以做流式细胞仪分析么?做流式细胞仪分析要用多少微升的悬液?谢谢!丁香网友zhaoyihenglk认为:一般我们使用500微升的悬液上机检测!!!问:请问流式检测的最小的细胞数量是多少,如果小于一个细胞的话是否就不能采用流式检测了。
流式要求的细胞的最小体积是多少,我最近收集了少量细胞团,30-50 微米,因为将细胞团中的各个细胞分离开较难,是否可以用于流式的检测?丁香网友XTYang认为:如果不能将细胞分散成单个细胞悬液的话是无法用流式细胞法分析的。
丁香网友swarm认为:检测时最少要有2000个细胞,但最初准备样品时,如果只做外标,样品比最少细胞数大一个数量级即可;但要是需要作内标的话,固定液和穿膜液粘度大,细胞损失也多,而且,穿膜液一般对细胞还有损伤,故样品细胞数要比最少细胞数大2个数量级!做流式,细胞必须是单细胞悬液,成团细胞可用胰酶处理,或是在细胞样品中加2mM的EDTA,防螯合。
问:请问流式检测的最小细胞数是多少?如分离出两种细胞,一种细胞数很少,但体积很大,另一种细胞是其数量的10-20倍,是否可以用流式同时检测呢?丁香网友XTYang认为:记得有人研究用针头取样后进行流式分析的文章,所以细胞数应该是可以很少的.在大量细胞中检测低频率事件(即你例子中的第一种细胞)正是流式的优点.关键是你的第一种细胞在大小,密度或荧光标记上和其他大量细胞有区别,并且细胞团能用酶解或机械方法处理成单细胞悬液.问:流式能检测含多种细胞的标本吗?细胞碎片影响流式检测效果吗?怎样在检测前去除细胞碎片?丁香网友swarm认为:能检测含多种细胞的标本因为流式本身能根据细胞的大小及细胞内容物颗粒大小(即所谓的流式仪所用的前向光和侧向光)来区别不同的细胞群而通常做研究时是荧光标记你感兴趣的亚群进行分析含有碎片会有一点点影响,但用空白样品(未作任何荧光标记的样本)选定你想研究的细胞群体时,即可排除细胞碎片,因为碎片所反映的前向光和侧向光和完整的细胞基本不相同。
2、Western和RT-PCR所需细胞数问:我是实验新手,拟在细胞上行WB和RT-PCR,由于实验基础太差,不知道细胞水平行WB和RT-PCR所需多少细胞,一般用多大的板子培养才能提足所需蛋白和RNA,请园子里的朋友们不吝指教,多谢!丁香网友zhangyuelin1999认为:我做过WB的,细胞数目要达到10*6个,这样提出的蛋白就可以了!因此一般的六孔板就可以了!24孔板,96孔的感觉有点小了,空间太小细胞长就不好了!先前要计个数的,然后进行蛋白定量的!悬浮的就直接离心得沉淀,裂解得蛋白的就可以了,贴壁的要用到细胞刮子。
RT-PCR 就不太清楚了,祝实验顺利!丁香网友everever认为:看你要养的细胞是哪种了,有的细胞的体积就很小,而有的细胞则铺得很开。
做RT-PCR用两步法即先抽RNA经反转录再做PCR一般要用到1ug的总RNA。
(1ug经反转录成20ul体系后一般可做至少50次20ul体系的PCR)常规做WB一个孔道上样要10*5个细胞。
但也和你的目的蛋白及提取的方式有关。
如果是膜蛋白或是某细胞器内分布的蛋白细胞数得增多。
你最好和你们实验室做过相关实验的人讨论一下。
问:我初次作RT-PCR 和Western,请教要使用多少转染后的细胞呢?转染效率是多少?瞬时转染是否可以传代呢?对检测有多大的影响?丁香网友lym569认为:一般转染效率达到50%的话,一个24孔班板的细胞足够做RT-PCR 和Western了,你可以做一个6孔板转染或2个24孔细胞转染.瞬时转染可以传代,但传代后所转染的细胞会越来越少.你传代的目的是什么?如果想稳定表达的话,你应该作稳定转染。
问:你的“一般转染效率达到50%的话,一个24孔班板的细胞足够做RT-PCR 和Western了”是不是可以这样理解:转染效率50%,24孔板上每个孔均长满细胞。
一半细胞用作RT-PCR ,另一半用作Western。
再弱弱的问:是刚好够吗?您的建议:你可以做一个6孔板转染或2个24孔细胞转染。
24孔板2个是为了保险。
那6孔板是??丁香网友lym569认为:转染效率50%,24孔板上每个孔均长满细胞。
一半细胞用作RT-PCR ,另一半用作Western 足够用了. 做2孔是其中一孔做RT-PCR ,另一孔做Western,这样操作起来比较方便,一个24孔板上的细胞够你做几次Western了.如果用6孔板,你只转染一个孔就可以,看你喜欢哪种了。
3、MTT细胞数问题问:本人准备做SKOV3和SMMC-7721的MTT实验,请问MTT实验中96孔板中的合适细胞数或者浓度。
丁香网友rfei8510认为:调整细胞浓度至1×105/ml,分别加入96孔板,每孔加200μL使每孔细胞数目是2×104个。
请教:各位在做MTT时,如何尽量保证各孔细胞数基本一致?混匀细胞所用容器是培养皿还是储液器?储液器用的是一次性的还是可重复使用的,哪家公司的,价格?丁香网友clearair00认为:MTT检测时各孔细胞数的均一可比是关键。
首先要保证收集细胞的分散混匀,贴壁细胞要消化完全,悬浮集落细胞团块要吹散充分,最好显微镜下观察确定。
其次微量取液操作要规范、稳定,建议选用精密度较高的微量移液器和质地优良的Tip头。
至于混匀细胞的场所是选择培养皿还是储液器,抑或其它容器,应该没有很大的影响,只要方便操作且不影响细胞活力即可灵活选用。
额外补充一句,MTT检测结果的影响因素除上面提及的方面外,每孔接种细胞的密度也是保证获取有意义和正确结果的重要一环,具体的接种细胞密度要根据不同的实验目的和观察效应的差异进行调整、确定。
请问做MTT时,96孔板中每孔的细胞数是多少啊?为了照相每次我都在加MTT前将扳子的培养液甩掉,照完相后加70微升的无血清培养液(为了节省)和20微升的MTT,不知道这样对结果有没有影响?丁香网友偷懒的猪认为:不同的细胞,不同的药物作用时间或是影响因素,要求接种的细胞数会不同。
一般是1000-10000个,看你的细胞的生长速度,体积大小,都会有影响的。
我做的几个不同的肿瘤细胞系,多数种3000-5000个,药物作用48-72小时是可以的,细胞数太少,读数太低,误差会增大。
细胞数太多,细胞长满了以后脱落,OD值和活细胞之间就不再是线性关系了,也会使结果失真。
我们这里也有人种1000/孔,但我试过2500/孔,作用48小时,OD值就特别小了。
MTT的影响因素太多了,还要自己好好摸索,愿你好运!我在用MTT法作细胞的生长曲线,现在遇到了几个问题,希望有人能帮我解决。
(1)OD值转换成细胞数的方法?(2)我得出的OD值都是在2-3之间,准确吗?(3)我不知道选择哪个波长490和570的,就选了两个?570的值比490的高,我该用哪个值?丁香网友ryochan 认为:(1)OD值转换为细胞数似乎不太好操作,就像楼上战友说的那样,影响因素比较多,单个细胞活性与MTT转化量是有关系的,所以需要有一个标准细胞活性的参照才能计算细胞数。
(2)OD应该在0.2~1之间线性度最好,2-3就不太好了,有时候<1.5也凑合了。
(3)标准的MTT,DMSO溶的话,吸光度峰值应该在570nm,线性度最高,但是多数实验室酶标仪的滤光片配置都没有这个数值,所以似乎大家就用490nm来替代了,理论上说线性度不如570nm,但是因为相差不算太多,也可以代用。
丁香网友忆歆人认为:可以先通过配制梯度浓度的细胞数,利用MTT法测OD值,将细胞数同OD值之间建立线性关系,即一标准曲线。
再根据标准曲线,通过OD 查找对应的细胞数!请教一下细胞数目是怎么调的,我的方法如下:先需7-8X104/ml的细胞浓度,把培养瓶中的细胞消化后,离心,弃去上清液,沉淀中加入少量培养基,制成细胞悬液,另取一预先消毒的大口瓶,加入培养基,所加培养基的量以超过自己所设计的96孔板上所需的接种量少许为宜,然后,用吸管吸取少量悬液,滴入大口瓶中,用玻璃计数板计数,四个大格平均数为7-8个,如果不够,继续滴加初始悬液,最终制成7-8X104/ml的细胞悬液。
刚开始做MTT,还不太熟,请大家指教。
不知道这种方法对不对?丁香网友hualey认为方法太复杂了,他的方法:正常消化,观察细胞明显收缩后,倒掉胰酶,用少许培养基轻轻荡洗细胞,弃去培养液,加入少许培养液吹打细胞,使之从壁上掉下,转移至离心管中用直头滴管吹打成单细胞,细胞计数,测得的密度除以所要求的密度,得到要稀释的倍数,稀释。
主要不同在于如果先稀释,可能会浪费培养基,至于其它操作,也可能是个人习惯问题,希望对你有帮助。
丁香网友jjw703 认为:调细胞浓度我们做的很多,方法和战友hualey的基本相同。
不同的是我们实验室在消毒细胞培养瓶的同时,还消毒一批小的瓶子,从10ml到20ml,这可以用来调细胞浓度。
具体方法是消化收集细胞,转入消毒好的小瓶子,然后细胞计数,得到一个原始浓度。
根据你所要的浓度的细胞悬液的量来稀释。
比如你调的原始细胞浓度是2.4×10 5/ml,需要最终细胞浓度8×10 4/ml 一共10ml,那么你就可以取原始细胞悬液3.3ml于另一个消毒好的小瓶子中,再补加6.7ml培养液。
我们的方法,一是可以节省离心管,而是可以节省培养基。
另外说个体外话,做MTT一定要细胞悬液浓度均匀,所以接种空板时,每加一排孔,都要混匀,把人为操作的误差减少到最低限度。
问:是否初始浓度的细胞,不用知道具体的体积?丁香网友wolfskin认为:(1)贴壁细胞胰酶消化,7-8ml培养基吹打呈单个细胞悬液;(2)取1.5ml离心管,PBS 450ul,细胞悬液50ul,即稀释10倍后计数;(3)计算密度,换算所需稀释倍数。
目前在养细胞,加不同刺激因素作用于细胞,通过MTT测OD值,观察刺激因素对细胞活力的影响。
但作了很多次,除第一次结果OD值均在1以下,其他都有1点几,2点几,有时甚至高达6.0(此时细胞加了MTT20UL/孔后,上清液就变成混浊紫色),而老板说数值太大,一般OD值应小于1。
我每次接种细胞密度大概有3-5x105个/ml,是否密度太大?不知各位战友作MTT数值一般多少?此外,为什么有很多次在我刚加了MTT后有些孔上清就变灰紫色,,抚育4h后更多孔上清变色,都看不清孔底的结晶了,是细胞活力太差还是MTT出现问题?丁香网友essie2003认为:(1)密度有点大了,你得OD值偏大应该和细胞数量关系很大,小于等于105左右比较合适。