细菌全基因组测序分享资料
- 格式:ppt
- 大小:1.85 MB
- 文档页数:17

全基因组重测序基础及⾼级分析知识汇总全基因组重测序是通过对已有参考序列(Reference Sequence)的物种的不同个体进⾏基因组测序,并以此为基础进⾏个体或群体⽔平的遗传差异性分析。
通过全基因组重测序,研究者可以找到⼤量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插⼊缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异位点。
基于以上变异位点作为分⼦遗传标记,在⼈类复杂疾病、动植物经济性状和育种研究及物种起源、驯化、群体历史动态等⽅⾯具有重⼤的指导意义(Bentley2006; Casillas& Barbadilla 2017)。
⼀、 基础理论知识全基因组重测序研究主要是依据在全基因组⽔平发现的分⼦遗传标记进⾏物种的群体遗传学研究,进⼀步的利⽤统计⽅法进⾏影响表型和经济性状候选基因和功能突变的研究。
分⼦群体遗传学研究的理论基础知识及统计分析⽅法⽇趋完善和呈现多样性,作为初学者,有必要对其中的⼀些基础概念有⼀定的了解,才能为后续的深⼊学习、研究提供基⽯。
以下基础知识主要参考国内动物遗传学书籍和最新的⼀篇关于分⼦群体遗传学⽅⾯的综述改变⽽成(吴仲贤编1961; 李宁2011; 吴常信2015; Casillas & Barbadilla 2017)。
⾼通量测序技术作为分⼦群体遗传学研究的有⼒⼯具,在科学研究、⽣产及疾病诊断治疗中起到原来越重要的作⽤,对关于⾼通量测序相关的理论基础知识进⾏⼀定程度的了解,也有助于⽂献阅读和。
1. 群体遗传学基础知识群体(Polulation):是指⽣活在⼀定空间范围内,能够相互交配并⽣育具有正常⽣殖能⼒后代的同种个体群。
等位基因频率(Alleles frequency):在⼀个群体中,某类等位基因占该基因位点上全部等位基因数的⽐率。

“Iontorrent微生物(细菌)全基因组重测序文库构建实验方案”清晨的阳光透过实验室的窗户,洒在略显拥挤的操作台上,我的思绪开始像电流一样流转,10年的方案写作经验让我对每一个细节都了如指掌。
就让我们一起探索这个关于Iontorrent微生物(细菌)全基因组重测序文库构建的实验方案吧。
我们要明确实验的目的:通过Iontorrent平台,对微生物(细菌)进行全基因组重测序,以获取更准确、更全面的基因组信息。
那么,就是具体的步骤了。
一、实验材料与仪器1.材料:微生物(细菌)样本、DNA提取试剂盒、磁珠、PCR试剂、测序试剂盒等。
2.仪器:Iontorrent测序仪、离心机、PCR仪、磁珠分离器、凝胶成像仪等。
二、实验步骤1.样本处理:将微生物(细菌)样本进行适当的预处理,如离心、洗涤等,确保样品的纯净度。
2.DNA提取:使用DNA提取试剂盒,按照说明书操作,提取微生物(细菌)的基因组DNA。
3.DNA定量:利用纳米滴技术或紫外可见分光光度计,对提取的DNA进行定量,以确保DNA的浓度和纯度。
4.DNA片段化:将提取的DNA进行片段化处理,使其成为适合测序的片段大小。
5.文库构建:将片段化的DNA与测序适配器连接,通过PCR扩增,构建测序文库。
6.文库质检:利用凝胶成像仪,对构建的文库进行质检,确保文库的质量。
7.测序:将质检合格的文库上机测序,使用Iontorrent测序仪进行测序。
8.数据分析:将测序得到的原始数据进行质控、比对、组装等分析,得到微生物(细菌)的全基因组序列。
9.结果验证:对测序结果进行验证,如通过PCR、一代测序等方法,确保测序结果的准确性。
三、注意事项1.实验过程中,要注意无菌操作,避免样本污染。
2.操作过程中,要严格按照说明书进行,确保实验的准确性。
3.测序数据的质量控制和分析是关键步骤,要密切关注每个环节,确保结果的可靠性。
4.实验过程中,要随时记录实验数据和操作过程,以便后续的数据整理和分析。

微生物基因组测序技术及其应用随着科技进步,微生物基因组测序技术在医学、环境、农业等领域受到广泛关注和应用。
本文将简要介绍微生物基因组测序技术的基本原理和应用场景,以及未来的发展方向。
一、微生物基因组测序技术的基本原理微生物基因组测序技术是指将微生物DNA分子逐一排列,从而得到一条由ATCG四种碱基构成的“基因序列”。
这种技术的基本原理是将DNA从细胞中分离出来,通过PCR扩增等方法得到大量的DNA片段,然后用高通量测序仪对这些DNA片段进行测序,最后将这些片段拼接得到完整的基因组序列。
目前,微生物基因组测序技术主要分为三种方法:Sanger测序技术、454逆转录聚合酶链反应测序技术和Illumina测序技术。
其中,Illumina测序技术是目前最常用的基因组测序方法之一。
二、微生物基因组测序技术的应用场景1.医学应用微生物基因组测序技术被广泛应用于临床诊断中。
如何对感染病原体进行准确快速的鉴定,是临床医生面临的一项困难。
传统的菌落培养法不仅时间长,而且不能鉴定细菌的种系,因此不能满足对临床诊断的要求。
微生物基因组测序技术可以直接从感染部位分离出细菌DNA,进行基因组测序后,通过对基因组序列的比对,快速高效地鉴定病原菌种类以及其耐药性。
同时,该技术还被应用于研究小肠细菌群的变化,对于小肠细菌感染和肠道菌群失调的诱因和机制的研究有着重要的作用。
而在抗生素的研究和开发中,微生物基因组测序技术也发挥着越来越重要的作用。
2.环境应用微生物基因组测序技术的应用不仅局限于医疗领域,也被广泛应用于环境监测领域。
通过微生物基因组测序技术,可以对环境中微生物丰度、多样性和功能进行高通量测定,揭示微生物群落结构和功能特征。
例如,饮用水中的微生物群落结构和数量分布对水质安全和人体健康有着至关重要的作用。
通过微生物基因组测序技术,可以对水中的细菌、病毒和病原真菌等微生物进行定量和定性分析,为水质监测提供有效的手段。
3.农业应用微生物基因组测序技术在农业领域的应用也越来越广泛。

细菌鉴定测序全文共四篇示例,供读者参考第一篇示例:细菌鉴定测序是一种通过测序细菌基因组DNA序列来确定细菌种类和特征的技术。
随着生物技术的发展和普及,细菌鉴定测序在医学、农业、环保等领域的应用越来越广泛,为研究人员提供了一种快速、准确的方法来识别细菌并了解其特性。
细菌是一类微生物,它们存在于地球上的各个环境中,包括土壤、水体、空气等。
细菌在生态系统中起着重要的作用,有些细菌对环境有益,有些则可能对人类和动植物造成危害。
正确鉴定细菌种类对于环境保护、疾病预防和治疗等方面具有重要意义。
传统的细菌鉴定方法包括形态学观察、生理生化试验和免疫学方法等,这些方法需要较长时间并且有一定的局限性。
而细菌鉴定测序技术则通过对细菌的DNA序列进行测序和分析,可以准确地确定细菌的种类和特征,同时还可以帮助研究人员深入了解细菌的生物学特性。
细菌鉴定测序的步骤主要包括DNA提取、测序反应、数据分析和结果解读等。
首先是DNA提取,通过将细菌培养物中的DNA提取出来,然后进行PCR扩增等处理来准备测序样本。
接着是测序反应,将DNA样本进行测序反应,得到细菌DNA的序列信息。
数据分析是整个过程中最关键的一步,通过对测序数据进行比对和分析,确定细菌的种类和特征。
最后是结果解读,根据数据分析的结果和相关数据库信息来判断细菌的鉴定结果。
细菌鉴定测序技术有许多优点,首先是快速性和准确性。
相比传统的鉴定方法,测序技术可以在较短的时间内完成细菌鉴定,而且结果更加可靠和准确。
其次是高通量性,测序技术可以同时对多个样本进行测序,大大提高了工作效率。
测序技术还可以帮助研究人员发现新的细菌种类和基因功能,对于科学研究具有重要的推动作用。
在医学领域,细菌鉴定测序技术被广泛应用于致病菌的鉴定和药物治疗方面。
通过对病原菌进行测序鉴定,可以为临床医生提供更准确的诊断结果和治疗方案,从而有效地控制传染病的传播。
在农业领域,细菌鉴定测序技术可以帮助农民鉴定土壤中的有益细菌,并设计出更加科学的农业生产方式。



全基因组测序与生物信息学分析在细菌耐药性研究中的应用一、本文概述随着全球抗生素耐药细菌的不断涌现,细菌耐药性问题已成为全球公共卫生领域面临的重大挑战。
全基因组测序(Whole Genome Sequencing,WGS)与生物信息学分析作为现代生物学研究的重要工具,在细菌耐药性研究中的应用日益凸显。
本文旨在探讨全基因组测序与生物信息学分析在细菌耐药性研究中的应用,以期为深入理解细菌耐药机制、发现新的耐药基因及开发新型抗菌药物提供理论支持和实践指导。
本文将简要介绍全基因组测序的基本原理及其在细菌耐药性研究中的应用,包括耐药基因的识别、耐药机制的分析以及耐药菌株的溯源等方面。
本文将重点阐述生物信息学分析在细菌耐药性研究中的作用,包括基因组序列的组装与注释、耐药基因的功能预测与验证、耐药网络的构建与分析等。
本文将展望全基因组测序与生物信息学分析在细菌耐药性研究中的未来发展趋势,包括耐药基因数据库的完善、耐药机制研究的深入以及新型抗菌药物研发的应用等。
通过本文的阐述,旨在提高读者对全基因组测序与生物信息学分析在细菌耐药性研究中的认识和理解,为相关领域的研究人员提供有益的参考和借鉴。
二、全基因组测序技术在细菌耐药性研究中的应用全基因组测序(Whole Genome Sequencing, WGS)技术,作为一种高分辨率的遗传分析工具,近年来在细菌耐药性研究中的应用日益广泛。
WGS技术能够一次性对细菌的全部基因进行深度测序,从而获取细菌的全基因组信息,包括耐药基因、毒力基因、基因变异等,为揭示细菌耐药的分子机制提供了有力支持。
WGS技术在细菌耐药性研究中的应用主要体现在以下几个方面:WGS能够快速准确地鉴定细菌种类和耐药基因。
通过比对全基因组序列,可以精确确定细菌的种属信息,同时识别出与耐药相关的基因或突变位点,从而明确细菌的耐药类型和耐药程度。
WGS有助于揭示细菌耐药的进化过程。
通过对不同时间点或不同来源的细菌样本进行WGS分析,可以追踪耐药基因的来源、传播和演化轨迹,揭示细菌耐药的进化动态和趋势。

Ion torrent微生物(细菌)全基因组重测序文库构建实验方案微生物全基因组重测序文库构建实验方案一、重测序原理全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
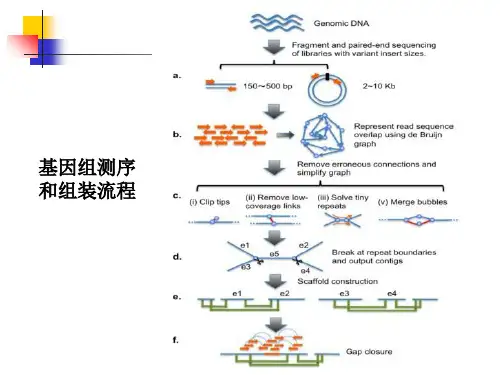
二、技术路线培养至对数期的单一菌落↓基因组DNA提取细菌DNA↓超声波打断 DNA片段化↓文库构建↓Ion OneTouch 乳液PCR、ES↓Ion PGM、Ion Proton 上机测序↓生物信息学分析DNA片段化↓末端修复↓纯化接头连接、缺口修复↓纯化文库片段筛选↓文库片段扩增↓纯化 Agilent Test、Qubit定量↓ Ion OneTouch System 重测序三、实验方案 1.细菌总DNA的提取液氮速冻、干冰保存的细菌菌液:若本实验室可以提供该细菌生长的条件,则对菌液进行活化,培养至对数期时,对该细菌进行DNA提取;若本实验室不能提供该细菌的生长条件,则应要求客户提供尽可能多的样本,以保证需要的DNA 量。
细菌DNA采用试剂盒提取法。
取对数生长期的菌液,按照细菌DNA提取试剂盒操作步骤进行操作。
提取完成后,对基因组DNA进行纯度和浓度的检测。
通过测定OD260/280,范围在之间则DNA较纯,使用Qubit对提取的DNA进行定量,确定提取的DNA浓度达到文库构建的量。
片段化采用Covaris System超声波打断仪,将待测DNA打断步骤:1)对待打断的DNA进行定量,将含量控制在100ng或者1μg2)打开Covaris M220安全盖,将Covaris AFA-grade Water充入水浴容器内,至液面到最高刻度线,软件界面显示为绿色3)将待打断DNA装入Ep LoBind管中,其中DNA为100ng 或1μg,加入Low TE至总体积为50mL4)将稀释的DNA转移至旋钮盖的Covaris管中,转移过程中不能将气泡带入,完成后旋紧盖子5)选择Ion_Torrent_200bp_50μL_ScrewCap_microTube,将对应的小管放入卡口,关上安全盖,点击软件界面“RUN”6)打断结束后,将混合液转移至一支新的离心管中3.末端修复及接头连接末端修复使用Ion Plus Fragment Kit进行,以100ng DNA量为例,各组分使用前瞬时离心2s 步骤:1)加入核酸酶free水至装有DNA片段的离心管中,至总体积为79μL 2)向体系中加入20μL 5×末端修复buffer,1μL末端修复酶,总体积为100μL 3)室温放置20min 片段纯化片段纯化使用Agencourt AMpure XP Kit进行步骤: 1)加入180μL Agencourt AMpure XP Reagent beads 于经过末端修复的离心管中,充分混匀,室温放置5min 2)瞬时离心后,将离心管放置于磁力架3min,至溶液澄清,小心去除上清,离心管保持放置在磁力架上3)离心管放置于磁力架上不移动,配置新的500μL 70%乙醇,加入,30s后,盖上盖子颠倒混匀2次,使磁珠悬浮,放回磁力架至溶液澄清后,小心除去上清 4)重复第三步5)用20μL墙头小心吸去多余的液体6)将离心管留在磁力架上,室温风干不超过5min7)取下离心管,加入25μL Low TE缓冲液,盖上盖子,上下颠倒混匀5次,旋窝震荡10s,充分混合8)瞬时离心后,将离心管放于磁力架上至少1min,溶液澄清后,将上清移入一支新的 EP管中 9)纯化的DNA片段用于下一步的接头连接4.接头连接、缺口修复和纯化片段两端的接头分别为barcode接头和P1接头接头连接在体系中加入 DNA 25μL 10×Ligase Buffer 10μL Ion P1 Adaptor 2μL Ion Xpress Barcode X+ 2μL dNTP Mix 2μL Nuclease-free water 49μL DNA Ligase 2μL Nick pair polymerase8μL Total 100μL将体系配置好后,放入PCR仪按照下面的温度进行1个循环扩增 25℃15min72℃5min 4℃hold 循环×1立即转移至一个新的离心管中纯化片段纯化使用Agencourt AMpure XP Kit进行步骤: 1)加入140μL Agencourt AMpure XP Reagent beads 于经过末端修复的离心管中,充分混匀,室温放置5min 2)瞬时离心后,将离心管放置于磁力架3min,至溶液澄清,小心去除上清,离心管保持放置在磁力架上3)离心管放置于磁力架上不移动,配置新的500μL 70%乙醇,加入,30s后,盖上盖子颠倒混匀2次,使磁珠悬浮,放回磁力架至溶液澄清后,小心除去上清 4)重复第三步5)瞬时离心,用20μL墙头小心吸去多余的液体 6)将离心管留在磁力架上,室温风干不超过5min7)取下离心管,加入20μL Low TE缓冲液,盖上盖子,上下颠倒混匀5次,旋窝震荡10s,充分混合8)瞬时离心后,将离心管放于磁力架上至少1min,溶液澄清后,将上清移入一支新的 EP管中5.文库片段筛选方案1E-Gel Agarose System采用E-Gel SizeSelect 2% Agarose Gel,在E-Gel电泳、成像系统中按照操作说明进行。

微生物全基因组序列数据的分析与注释随着生物技术的不断发展,微生物全基因组序列数据的获取和分析变得越来越容易,已经成为微生物学研究的一项重要工作。
但是,如何对这些大量数据进行正确的分析和注释,以及如何从中挖掘出有效的信息,仍然是微生物学家们需要解决的问题。
本文将从以下几个方面介绍微生物全基因组序列数据的分析与注释。
一、全基因组序列数据分析的流程1. 数据准备首先需要对采集到的原始数据进行处理,包括质控、去除低质量序列、剔除可能的污染物等,以得到高质量的序列数据。
2. 基因组组装接下来需要对序列进行组装,将得到的短序列拼接成较长的连续序列,建立起基因组的局部和整体结构。
3. 基因预测与注释利用相应的软件对基因组序列进行预测和注释,将可能存在的编码蛋白序列识别出来,并对不同的基因进行分类、注释,以及进行功能预测。
4. 基因组比较通过将已知的基因组与样本进行比较,找出基因组中存在的差异、重复、插入、缺失、基因家族和同源关系等信息。
5. 基因表达分析通过将RNA测序和基因组序列比较,可以分析出基因的表达模式和水平,以及相关的基因调控因子。
二、全基因组序列数据注释的方法基因注释是将基因组序列与已有数据库中的信息进行比较,以识别和确定序列的生物学含义和功能的过程。
1. Blast(基于比对的注释方法)Blast是最常见的基因组注释方法之一。
通过将基因组序列比对到已有的数据库中,找到最相似的基因,从而确定基因的功能。
2. GO注释(基于功能分类的注释方法)GO(Gene Ontology)是一套用于描述基因和其功能的标准化系统。
通过将基因功能与GO系统中现有的注释信息进行比对,确定基因的分子功能类型和生物学过程。
3. KEGG注释(基于通路分析的注释方法)KEGG(Kyoto Encyclopedia of Genes and Genomes)是一个描述生物通路的数据库。
将基因组注释结果与KEGG数据库比对,可以确定基因参与的代谢通路和信号通路等信息。
