PGM测序方法rotocol
- 格式:docx
- 大小:644.03 KB
- 文档页数:16
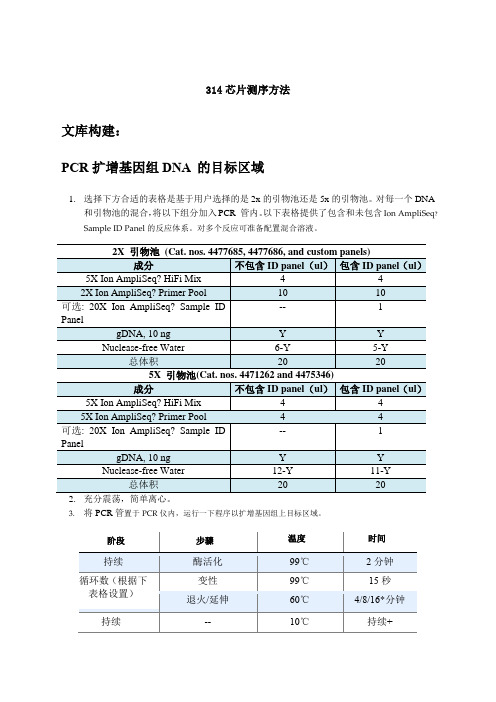
314芯片测序方法文库构建:PCR扩增基因组DNA 的目标区域1.选择下方合适的表格是基于用户选择的是2x的引物池还是5x的引物池。
对每一个DNA和引物池的混合,将以下组分加入PCR 管内。
以下表格提供了包含和未包含Ion AmpliSeq?Sample ID Panel的反应体系。
对多个反应可准备配置混合溶液。
2.充分震荡,简单离心。
3.将PCR管置于PCR仪内,运行一下程序以扩增基因组上目标区域。
*4分钟是对≤1536对引物池;8分钟是对1537-6144对引物池;16分钟是对6145-24576对引物池。
+PCR产物可置于10℃过夜储存。
注意:Ready‐to‐use panels: 当加入Ion AmpliSeq? Sample ID panel时不需要调整循环数。
Custom primer pools:如果一个custom 设计的panel中包含两个引物池,并分别落在不同的循环数分类中,请选用更大的循环数来扩增这两个引物池。
一般标准样品可以增加一个循环,从而保证其扩增效率,石蜡切片样品可增加2-3个循环。
如果引物池有两管及以上,应当分别扩增(10μl体系)。
扩增完后混合取20μl进行下轮实验。
暂停点– PCR产物可以储存在10°C过夜,需要更长时间储存,可以置于–20°C.部分消化引物序列1.轻度离心PCR管将反应后溶液收集到管底。
往每个混合反应液中加入2微升FuPa?Reagent?,总体系达到22微升。
2.充分振荡混匀,轻度离心将液体收集到管底。
(也可以选择用枪吸取一半以上的液体上下吹打至少5次来混匀)3.将PCR管放入PCR仪,按以下程序运行。
将接头连接至扩增产物并纯化配置并运行连接反应1.如果有可见的沉淀物在Switch溶液中,通过室温下震荡或上下吹吸来溶解并重悬。
2.小心PCR管加入以下成分到每个反应孔内消化样本。
在加入之前可先混合Switch溶液和接头。


高通量测序常用名词汇总一代测序技术:即传统的Sanger测序法,Sanger法是根据核苷酸在待定序列模板上的引物点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以A、T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸dNTP,并混入限量的一种不同的双脱氧核苷三磷酸ddNTP;由于ddNTP缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T 或C处终止,使反应得到一组长几百至几千碱基的链终止产物;它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,通过检测得到DNA碱基序列;二代测序技术:next generation sequencingNGS又称为高通量测序技术,与传统测序相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定,从而使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序Deep sequencing;NGS主要的平台有Roche454 & 454+,IlluminaHiSeq 2000/2500、GA IIx、MiSeq,ABI SOLiD等;基因:Gene,是遗传的物质基础,是DNA或RNA分子上具有遗传信息的特定核苷酸序列;基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状;DNA:Deoxyribonucleic acid,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱基、脱氧核糖、磷酸;脱氧核糖核酸通过3',5'-磷酸二酯键按一定的顺序彼此相连构成长链,即DNA链,DNA链上特定的核苷酸序列包含有生物的遗传信息,是绝大部分生物遗传信息的载体;RNA:Ribonucleic Acid,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成;核糖核苷酸经磷酯键缩合而成长链状分子称之为RNA链;RNA是存在于生物细胞以及部分病毒、类病毒中的遗传信息载体;不同种类的RNA链长不同,行使各式各样的生物功能,如参与蛋白质生物合成的RNA有信使RNA、转移RNA和核糖体RNA等;16S rDNA:"S"是沉降系数,是反映生物大分子在离心场中向下沉降速度的一个指标,值越高,说明分子越大;rDNAribosome DNA指的是原核生物基因组中编码核糖体RNArRNA分子对应的DNA序列,16S rDNA 是原核生物编码核糖体小亚基16S rRNA的基因;细菌rRNA核糖体RNA按沉降系数分为3种,分别为5S、16S和23S rRNA;16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列,存在于所有细菌染色体基因中;16S rRNA 普遍存在于原核生物中;16S rRNA 分子,其大小约1540bp,既含有高度保守的序列区域,又有中度保守和高度变化的序列区域,其可变区序列因细菌不同而异,恒定区序列基本保守,所以可利用恒定区序列设计引物,将16S rDNA片段扩增出来,通过高通量测序利用可变区序列的差异来对不同菌属、菌种的细菌进行分类鉴定;cDNA:complementary DNA,互补脱氧核糖核酸,与RNA链互补的单链DNA,以RNA为模板,在反转录酶的作用下所合成的DNA;Small RNA:生物体内一类高度保守的重要的功能分子,其大小在18-30nt,包括microRNA、siRNA、snRNA、snoRNA和piRNApiwi-interacting RNA等,它的主要功能是诱导基因沉默,调控细胞生长、发育、基因转录和翻译等生物学过程;以miRNA为例介绍它们的功能:miRNA 与RNA诱导沉默复合体RNA induced silencing complex, RISC结合,并将此复合体与其互补的mRNA序列结合,根据靶序列与miRNA的互补程度,从而导致靶序列降解或干扰靶序列蛋白质的翻译过程;SD 区域:Segment duplication,串联重复是由序列相近的一些 DNA 片段串联组成;串联重复在人类基因多样性的灵长类基因中发挥重要作用;Genotype and phenotype:基因型与表型,基因型是指某一生物个体全部基因组合的总称;表型,又称性状,是基因型和环境共同作用的结果;基因组:Genome,单倍体细胞核、细胞器线粒体、叶绿体或病毒粒子所含的全部DNA分子或RNA分子;全基因组de novo测序:又称从头测序,它不依赖于任何现有的序列资料,而直接对某个物种的基因组进行测序,然后利用生物信息学分析手段对序列进行拼接、组装,从而获得该物种的基因组序列图谱;全基因组重测序:对已有参考序列Reference Sequence物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的遗传差异性分析;全基因组重测序能够发现大量的单核苷酸多态性位点SNP、拷贝数变异Copy Number Variation,CNV、插入缺失InDel,Insertion/Deletion、结构变异Structure Variation,SV等变异类型,以准确快速的方法将单个参考基因组信息上升为群体遗传特征;转录组:Transcriptome,是指特定生长阶段某组织或细胞内所有转录产物的集合;狭义上指所有mRNA的集合;转录组测序:对某组织在某一功能状态下所能转录出来的所有RNA进行测序,获得特定状态下的该物种的几乎所有转录本序列信息;通常转录组测序是指对mRNA进行测序获得相关序列的过程;其根据所研究物种是否有参考基因组序列分为转录组de novo测序无参考基因组序列和转录组重测序有参考基因组序列;外显子组:Exome,人类基因组全部外显子区域的集合称为外显子组,是基因中重要的编码蛋白的部分,并涵盖了与个体表型相关的大部分的功能性变异;外显子组测序:是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法;外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、InDel 等具有较大的优势;目标区域测序:应用相关试剂盒对基因组上感兴趣的目标区域进行捕获富集后进行大规模测序,一般需要根据目标区域专门定制捕获芯片;宏基因组:Metagenome,指特定生活环境中全部微小生物遗传物质的总和;它包含了可培养的和未可培养的微生物的基因;目前主要指环境样品中的细菌和真菌的基因组总和;宏基因组16S rRNA测序:可以对特定环境下的细菌和古细菌群体的微生物种类和风度进行有效的鉴定;对不同地点、不同条件下的多个样本16S rRNA的PCR产物平行测序,可以比较不同样本间的微生物组成及成分差异,进而阐明物种丰度、种群结果等生态学信息;表观遗传学:Epigenetics,是指在基因组DNA序列没有改变的情况下,基因的表达调控和性状发生了可遗传的变化;表观遗传的现象很多,已知的有DNA甲基化DNA methylation,基因组印记genomic impriting,母体效应maternal effects,基因沉默gene silencing,核仁显性,休眠转座子激活和RNA编辑RNA editing等;全基因组甲基化测序:DNA 甲基化是指在 DNA 甲基化转移酶的作用下,在基因组 CpG 二核苷酸的胞嘧啶5'碳位共价键结合一个甲基基团;DNA 甲基化已经成为表观遗传学和表观基因组学的重要研究内容;甲基化是基因表达的主要调控方式之一,研究染色体DNA甲基化情况是了解基因调控的重要手段;对已经有参考基因组的物种的基因组DNA用标准亚硫酸氢盐Bisulfite处理后,未甲基化的胞嘧啶C会脱氨基形成尿嘧啶U,经PCR扩增,U替换为胸腺嘧啶T,而发生甲基化的胞嘧啶C保持不变;将处理组与参考基因组序列进行比对,可发现甲基化位点并对甲基化情况进行定量分析的方法叫做全基因组甲基化测序;ChIp-Seq:Chromatin Immunoprecipitation sequencing,即染色质免疫共沉淀-测序技术,即通过染色质免疫共沉淀技术特异性地富集目的蛋白结合的DNA片段;对富集得到的DNA片段进行纯化与文库构建,然后进行高通量测序,从而得到全基因组范围内可以与目的蛋白相互作用的DNA片段的方法叫做ChIP-Seq;数字表达谱:Digital Gene Expression Profile,利用新一代高通量测序技术和高性能计算分析技术,能够全面、经济、快速地检测某一物种特定组织在特定状态下的基因表达情况,即运用特定的酶对mRNA距polyA tail 21-25nt的位置进行酶切,所获得的带polyA尾的序列Tag通过高通量测序,该tag被测得的次数即是对应基因的表达值;数字基因表达谱已被广泛应用于基础科学研究、医学研究和药物研发等领域;特点是经济,但获得的数据量有限;若想获得转录本的更多信息的话,一般都采用转录组测序的方法来测序;SBS:sequencing by synthesis,边合成边测序反应,是指在DNA聚合酶的作用下延伸碱基所进行的测序;Run:指高通量测序平台单次上机测序反应;图1. Flow Cell结构示意图Lane:也叫channel,单泳道,每条泳道包含2列column,每列分布有多个小区tile,如图1;不同的测序平台Flow Cell中所含的Lane不一样,如HiSeq 2000是2个flow cell,每个flow cell中含有8个lane;HiSeq 2500是包含2个mini flow cell快速运行模式和2个high output flow cell,两个模式不能同时运行,其中每个mini flow cell包含2个lane,每个high output flow cell中包含8个lane;Miseq系统的flow cell仅含有1个lane; Tile:小区,每条Lane中有2列tile,合计120个小区;每个小区上分布数目繁多的簇结合位点,如图1;Cluster:簇,在Illumina测序平台中会采用桥式PCR方式生产DNA簇,每个DNA簇才能产生亮度达到CCD可以分辨的荧光点;Index:标签,在Illumina平台的多重测序Multiplexed Sequencing过程中会使用Index 来区分样品,并在常规测序完成后,针对Index部分额外进行7个循环的测序,通过Index的识别,可以在1条Lane中区分12种不同的样品;Barcode:与Index同义,多指在Roche GS FLX 454测序平台的16S PCR产物的测序过程中接头序列所包含的的用来区分不同样本的序列;PF%:PF%是指符合测序质量标准的簇的百分比,与测序的通量相关联;Fasta:一种序列存储格式;一个序列文件若以FASTA格式存储,则每一条序列的第一行以“>”开头,而跟随“>”的是序列的ID号即唯一的标识符及对该序列的描述信息;第二行开始是序列内容,序列短于61nt的,则一行排列完;序列长于61nt的,则每行存储61nt,最后剩下小于61nt的,在最后一行排列完;第二条序列另起一行,仍然由“>”和序列的ID号开始,以此类推;Fastq:Fastq是Solexa测序技术中一种反映测序序列的碱基质量的文件格式;第一行以“”符号开头,后面紧跟一个序列的描述信息;第二行是该序列的内容;第三行以“+”符号开头,后面可以是该序列的描述信息,也可省略;而第四行是第二行中的序列内容每个碱基所对应的测序质量值;Read:高通量测序平台产生的序列标签就称为 reads;基因组组装:进行基因组或转录组de novo测序时,物种基因组经构建不同的文库测序所得的片段需经过生物信息学手段对其进行整理拼接,并通过一定的标准如N50对后续组装结果进行质量评估等,最终获得高准确度的基因组序列的过程;基因组测序深度:测序得到的总碱基数与待测基因组大小的比值;如测一个物种的全基因组的重测序,基因组大小约为5G,测序获得100G的数据量,则测序深度为20×;基因组覆盖率:指测序获得的序列占整个基因组的比例;由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap;例如一个细菌基因组测序,覆盖率是98%,那么还有2%的序列区域是没有通过测序获得的;Contig:在de novo测序中拼接软件基于 reads 之间的 overlap 区,拼接获得的中间没有gap的序列称为 Contig重叠群;Scaffold:基因组 de novo 测序,通过 reads 拼接获得 Contigs 后,往往还需要构建 454 Paired-end 库或 Illumina Mate-pair 库,以获得一定大小片段如 3Kb、8Kb、10Kb、20Kb 两端的序列;基于这些序列,可以确定一些Contig 之间的顺序关系,这些先后顺序已知的Contigs 组成 Scaffold;Contig N50:Reads拼接后会获得一些不同长度的Contigs;将所有的Contig长度相加,能获得一个Contig总长度;然后将所有的Contigs按照从长到短进行排序,如获得Contig 1,Contig 2,Contig 3……Contig 25;将Contig按照这个顺序依次相加,当相加的长度达到Contig总长度的一半时,最后一个加上的Contig长度即为Contig N50;举例:Contig 1+Contig 2+ Contig 3 +Contig 4=Contig总长度1/2时,Contig 4的长度即为Contig N50;Contig N50可以作为基因组拼接的结果好坏的一个判断标准;Scaffold N50:Scaffold N50与Contig N50的定义类似;Contigs拼接组装获得一些不同长度的Scaffolds;将所有的Scaffold长度相加,能获得一个Scaffold总长度;然后将所有的Scaffolds按照从长到短进行排序,如获得Scaffold 1,Scaffold 2,Scaffold 3……Scaffold 25;将Scaffold按照这个顺序依次相加,当相加的长度达到Scaffold总长度的一半时,最后一个加上的Scaffold长度即为Scaffold N50;举例:Scaffold 1+Scaffold 2+ Scaffold 3 +Scaffold 4 +Scaffold 5=Scaffold总长度1/2时,Scaffold 5的长度即为Scaffold N50;Scaffold N50可以作为基因组拼接的结果好坏的一个判断标准;Isotig:指在转录组de novo测序时,用454平台测序完成后组装出的结果,一个isotig可视为一个转录本;Isogroup:指转录组de novo测序中,用454平台测序完成后组装出的结果获得的可聚类到同一个基因的转录本群;GC%:GC含量,全基因组范围内或在特定基因组序列内的4种碱基中,鸟嘌呤和胞嘧啶所占的比率;SNP:single nucleotide polymorphism,单核苷酸多态性,个体间基因组DNA序列同一位置单个核苷酸变异替代、插入或缺失所引起的多态性;不同物种个体基因组 DNA 序列同一位置上的单个核苷酸存在差别的现象;有这种差别的基因座、DNA序列等可作为基因组作图的标志;SNP 在CG序列上出现最为频繁,而且多是C转换为T ,原因是CG中的C 常为甲基化的,自发地脱氨后即成为胸腺嘧啶;一般而言,SNP 是指变异频率大于 1 %的单核苷酸变异,主要用于高危群体的发现、疾病相关基因的鉴定、药物的设计和测试以及生物学的基础研究等;InDel:Insertion/Deletion,插入/缺失,在基因组重测序进行mapping时,进行容Gap的比对并检测可信的Short InDel,如基因组上小片段>50bp的插入或缺失;在检测过程中,Gap 的长度为1~5个碱基;CNV:copy number variation,基因组拷贝数变异,是基因组变异的一种形式,通常使基因组中大片段的DNA形成非正常的拷贝数量;如人类正常染色体拷贝数是2,有些染色体区域拷贝数变成1或3,这样,该区域发生拷贝数缺失或增加,位于该区域内的基因表达量也会受到影响;如果把一条染色体分成A-B-C-D四个区域,则A-B-C-C-D/A-C-B-C-D/A-C-C-B-C-D/A-B-D 分别发生了C区域的扩增及缺失,扩增的位置可以是连续扩增如 A-B-C-C-D 也可以是在其他位置的扩增,如A-C-B-C-D;SV:structure variation,基因组结构变异,染色体结构变异是指在染色体上发生了大片段的变异;主要包括染色体大片段的插入和缺失引起 CNV 的变化,染色体内部的某块区域发生重复复制、翻转颠换、易位、两条染色体之间发生重组inter-chromosome trans-location 等;基因表达差异:是指某一物种或特定细胞在特定时期/功能状态下,多样本间不同基因在mRNA水平上表达量的差异,可通过RPKM/FPKM值来体现;RPKM:Reads Per Kilobase per Million mapped reads Mortazavi etal., 2008,是指每 1 百万个map 上的reads 中map 到外显子的每1K 个碱基上的reads 个数;计算公式四RPKM=106C/NL/103,其中C为唯一比对到目的基因的reads数;N为唯一比对到参考基因的总reads数,L是目的基因编码区的碱基数;RPKM法可以消除基因长度、数据量之间的差异进行计算基因表达量;可变剪切:alternative splicing大多数真核基因转录产生的mRNA前体是按一种方式剪接产生出一种mRNA,因而只产生一种蛋白质;但有些基因产生的mRNA前体可按不同的方式剪接,产生出两种或更多种mRNA,即可变剪接;基因融合:Gene fusion,将基因组位置不同的两个或多个基因中的一部分或全部整合到一起,形成新的基因,称作融合基因或嵌合体基因,该基因有可能翻译出融合或嵌合体蛋白;基因家族分析:通过进行BLASTN/ HMM比对等查找基因归属的基因家族并添加相关功能注释;基因组注释:Genome annotation是利用生物信息学方法和工具,对基因组所有基因的生物学功能进行高通量注释,是当前功能基因组学研究的一个热点;基因组注释的研究内容包括基因识别和基因功能注释两个方面;基因识别的核心是确定全基因组序列中所有基因的确切位置;常见的基因组注释有GO注释、pathway分析;GO注释:gene ontology是指对基因功能的注解;GO强调基因产物在细胞中的功能;GO不能反映此基因的表达情况,即是否在特定细胞中、特定组织中、特定发育阶段或与某种疾病相关,但GO支持其他的OBOopen biology ontologies成员成立其他类型的本体论数据库如发育本体学、蛋白组本体学、基因芯片本体学等Pathway注释:是指对功能基因参与的信号通路等进行分析注释;甲基化率:是指在甲基化测序中,发生甲基化的胞嘧啶占所有胞嘧啶的比率;CpG岛:CpG island 是指DNA上一个区域,此区域含有大量相联的胞嘧啶C、鸟嘌呤G,以及使两者相连的磷酸酯键p;基因组中长度为300~3000 bp的富含CpG二核苷酸的一些区域,主要存在于基因的5’区域;启动子区中CpG岛的未甲基化状态是基因转录所必需的,而CpG序列中的C的甲基化可导致基因转录被抑制;Q20,Q30:基因的二代测序中,每测一个碱基会给出一个相应的质量值,这个质量值是衡量测序准确度的;碱基的质量值13,错误率为5%,20的错误率为1%,30的错误率为0.1%;行业中Q20与Q30则表示质量值≧20或30的碱基所占百分比;例如一共测了1G的数据量,其中有0.9G的碱基质量值大于或等于20,那么Q20则为90%;Q20值是指的测序过程碱基识别Base Calling过程中,对所识别的碱基给出的错误概率;质量值是Q20,则错误识别的概率是1%,即错误率1%,或者正确率是99%;质量值是Q30,则错误识别的概率是0.1%,即错误率0.1%,或者正确率是99.9%;质量值是Q40,则错误识别的概率是0.01%,即错误率0.01%,或者正确率是99.99%;全基因组测序全基因组测序-技术路线提取基因组DNA,然后随机打断,电泳回收所需长度的DNA片段0.2~5Kb,加上接头, 进行基因簇cluster制备或电子扩增E-PCR,最后利用Paired-EndSolexa或者Mate-PairSOLiD的方法对插入片段进行测序;然后对测得的序列组装成Contig,通过Paired-End的距离可进一步组装成Scaffold,进而可组装成染色体等;组装效果与测序深度与覆盖度、测序质量等有关;常用的组装有:SOAPdenovo、Trimity、Abyss等;全基因组测序-原理双末端Paired-End测序原理测序深度Sequencing Depth:测序得到的碱基总量bp与基因组大小Genome的比值,它是评价测序量的指标之一;测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降;重测序的个体,如果采用的是Paired-End 或Mate-Pair方案,当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证;测序深度对基因组覆盖度和测序错误率的影响HOM:纯合体 HET:杂合体全基因组测序-分析流程1.数据量产出总碱基数量、Totally mapped reads、Uniquely mapped reads统计,测序深度分析;2.一致性序列组装与参考基因组序列Reference genome sequence的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列;3.SNP检测及在基因组中的分布提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集;并根据参考基因组序列对检测到的变异进行注释; 4.InDel检测及在基因组的分布在进行mapping的过程中,进行容Gap的比对并检测可信的Short InDel;在检测过程中,Gap 的长度为1~5个碱基;5.Structure Variation检测及在基因组中的分布目前SBC能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等;根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进进行注释;全基因组重测序生物信息学分析流程。

焦磷酸测序技术流程Next-generation sequencing (NGS) has revolutionized the field of genomics, enabling researchers to sequence DNA at an unprecedented scale and speed. Among the various NGS techniques, pyrosequencing, also known as 454 sequencing or the 454 pyrosequencing method, has emerged as a powerful tool for DNA sequencing. In this article, we will delveinto the workflow of pyrosequencing, highlighting its key steps and providing a comprehensive understanding of this technique.Pyrosequencing begins with the preparation of a DNA library, which involves fragmenting the DNA into smaller pieces and attaching specific adapters to the ends of the fragments. These adapters serve as priming sites for subsequent amplification and sequencing reactions. Once the library is prepared, it is loaded onto a picotiter plate, which contains millions of tiny wells, each capable of accommodating a single DNA fragment.The next step in the pyrosequencing workflow is the emulsion PCR (polymerase chain reaction). Emulsion PCR is a method that allows for the amplification of individual DNA fragments within the wells of the picotiter plate. The DNA fragments, along with specific primers and PCR reagents, are encapsulated in water-in-oil emulsion droplets. These droplets serve as microreactors, each containing a single DNA fragment and all the necessary components for PCR amplification. By subjecting the emulsion to thermal cycling, multiple rounds of PCR amplification occur, resulting in the generation of clonal DNA clusters.Following emulsion PCR, the picotiter plate is loaded onto a sequencing instrument, such as the Roche 454 Genome Sequencer. The sequencing instrument utilizes a series of enzymatic reactions to generate light signals that correspond to the order of nucleotides in the DNA template. The sequencing process begins by introducing a single type of nucleotide (A, T, C, or G) into the reaction mixture. If the introduced nucleotide is complementary to the next nucleotide in the DNA template, a pyrophosphate molecule is released, which in turn generates a light signal. The lightsignal is detected by a CCD camera, and the nucleotide incorporation event is recorded as a peak in a pyrogram.After the detection of the nucleotide incorporation event, the enzyme responsible for generating the light signal is inactivated, and the signal is recorded. The released pyrophosphate is then enzymatically converted into ATP, which is subsequently degraded by a luciferase enzyme. The degradation of ATP generates a light signal that is proportional to the number of incorporated nucleotides. This process is repeated for each of the four nucleotides, allowing for the sequential determination of the DNA sequence.Once the sequencing run is complete, the raw data generated by the instrument is processed and analyzed using bioinformatics tools. This involves base calling, which converts the light signals into nucleotide sequences, and quality control, which assesses the reliability of the generated sequences. The resulting sequences can then be aligned to a reference genome or assembled de novo to reconstruct the original DNA sequence.In summary, pyrosequencing is a powerful and versatile DNA sequencing technique that allows for the rapid and accurate determination of DNA sequences. Its workflow encompasses steps such as DNA library preparation, emulsion PCR, nucleotide incorporation detection, and data analysis. With its high throughput and ability to generate long reads, pyrosequencing has found applications in various fields, including genomics, transcriptomics, and metagenomics, contributing to our understanding of the genetic basis of life.。

生物组学和基因测序的技术和方法人类的DNA编码了我们所拥有的全部遗传信息。
而通过生物组学和基因测序的技术和方法,人们可以更好地理解这些遗传信息,并应用于医学、农业、环保等领域。
生物组学是组织、细胞和DNA分析的学问。
其中,DNA测序是生物组学的重要组成部分。
DNA测序技术使用不同的发光、放射和电化学技术,读取DNA的不同区域,并生成基因序列数据。
而随着科技的进步,DNA测序技术的精度和速度都得到了显著提高。
第一代测序技术,包括了盖尔基(Sanger)测序和马克斯姆·吉尔伯特(Maxam-Gilbert)测序。
这些技术仍然被广泛应用于研究和诊断,但需要昂贵的仪器和大量时间和人力投入。
第二代测序技术的引入,改变了DNA测序的现状,这些技术包括了:胶体电泳测序、高通量法、Illumina测序和454测序。
这些技术提高了测序速度和精度,并显着降低了成本。
除了DNA测序,第二代测序技术也可用于RNA测序、表观基因组学、染色体结构和复合体的研究。
第三代测序技术的出现,带来了更高的速度和更低的成本。
这些技术包括Oxford Nanopore和PacBio。
它们的原理是通过监测DNA或RNA样本中的离子流或光学信号,直接读取单个分子序列,从而实现更加高精度的测序。
DNA测序技术的不断提高,使得人们可以获得更广泛的生物信息,也为医学研究和精准医疗提供了更多的信息来源。
例如,全基因组测序可以用于发现染色体异常、某些疾病基因变异。
基因甲基化测序可以用于研究表观遗传学相关疾病,如患有乳腺癌的女性,基因甲基化发生改变的可能性较高。
转录组测序可以用于研究基因表达的变化以及复杂的信号通路。
蛋白质组测序可以用于研究蛋白质结构和功能,及其与疾病的关系。
在生物组学领域,也有一项称为CRISPR-Cas9的技术引起了广泛的关注。
CRISPR-Cas9是一种基因编辑技术,可以用于切割、插入或删除DNA序列,以改变遗传信息。
这项技术被认为具有无限的潜力,可用于治疗遗传性疾病,或增强病毒对癌细胞的杀伤力。

314芯片测序方法文库构建:PCR扩增基因组DNA 的目标区域1.选择下方合适的表格是基于用户选择的是2x的引物池还是5x的引物池。
对每一个DNA和引物池的混合,将以下组分加入PCR 管内。
以下表格提供了包含和未包含Ion AmpliSeq™Sample ID Panel的反应体系。
对多个反应可准备配置混合溶液。
2.充分震荡,简单离心。
3.将PCR管置于PCR仪内,运行一下程序以扩增基因组上目标区域。
*4分钟是对≤1536对引物池;8分钟是对1537-6144对引物池;16分钟是对6145-24576对引物池。
+PCR产物可置于10℃过夜储存。
注意:Ready‐to‐use panels: 当加入Ion AmpliSeq™ Sample ID panel时不需要调整循环数。
Custom primer pools:如果一个custom 设计的panel中包含两个引物池,并分别落在不同的循环数分类中,请选用更大的循环数来扩增这两个引物池。
一般标准样品可以增加一个循环,从而保证其扩增效率,石蜡切片样品可增加2-3个循环。
如果引物池有两管及以上,应当分别扩增(10μl体系)。
扩增完后混合取20μl进行下轮实验。
暂停点– PCR产物可以储存在10°C过夜,需要更长时间储存,可以置于–20°C.部分消化引物序列1.轻度离心PCR管将反应后溶液收集到管底。
往每个混合反应液中加入2微升FuPa Reagent ,总体系达到22微升。
2.充分振荡混匀,轻度离心将液体收集到管底。
(也可以选择用枪吸取一半以上的液体上下吹打至少5次来混匀)3.将PCR管放入PCR仪,按以下程序运行。
将接头连接至扩增产物并纯化配置并运行连接反应1.如果有可见的沉淀物在Switch溶液中,通过室温下震荡或上下吹吸来溶解并重悬。
2.小心PCR管加入以下成分到每个反应孔内消化样本。
在加入之前可先混合Switch溶液和接头。
全基因组测序建库类型一、建库方法全基因组测序建库的方法主要分为两类:鸟枪法(Shotgun)和全长建库法(fosmid library)。
1. 鸟枪法(Shotgun):这种方法是先对基因组进行随机切割,然后对切割后的片段进行测序。
这种方法简单快速,但产生的数据质量相对较低,且对基因组的覆盖度较低。
2. 全长建库法(fosmid library):这种方法是将基因组的全长DNA片段插入到fosmid载体中,然后对插入后的fosmid进行测序。
这种方法产生的数据质量较高,且对基因组的覆盖度也较高。
二、测序策略全基因组测序的测序策略主要分为两种:单一测序(Single-ended sequencing)和双端测序(Paired-ended sequencing)。
1. 单一测序(Single-ended sequencing):这种方法只对一条DNA 链进行测序,通常用于鸟枪法建库的数据测序。
2. 双端测序(Paired-ended sequencing):这种方法对两条DNA链进行测序,通常用于全长建库法的数据测序。
双端测序可以得到更多关于基因组结构的信息,如基因、重复序列等。
三、测序平台目前全基因组测序的测序平台主要是基于下一代测序技术(Next Generation Sequencing,NGS)。
主要的测序平台包括:1. Illumina平台:这是目前应用最广泛的NGS平台,具有高通量、高精度和高速度等优点。
2. SOLiD平台:这是另一种NGS平台,具有双端测序能力,可以提供更多的基因组信息。
3. 454平台:这是基于焦磷酸测序技术的平台,具有读长较长的优点,但通量相对较低。
4. PacBio平台:这是基于单分子实时测序技术的平台,具有无需进行PCR扩增等优点,但数据质量受限于测序模板的质量。
四、数据处理全基因组测序的数据处理主要包括以下步骤:1. 序列质量控制:包括去除低质量序列、去除序列中的污染等。
高通量测序领域常用名词解释大全什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
高通量测序技术及其应用随着科学技术的不断进步,人类对基因组学的了解越来越深入。
高通量测序技术作为基因组学领域的一项重要技术,已经成为基因研究的利器之一。
本文将为您介绍高通量测序技术的原理和应用。
一、高通量测序技术的原理高通量测序技术是指利用高通量平台进行大规模的DNA或RNA测序,其过程主要包括文库构建、序列生成和数据分析三个部分。
文库构建是指将待测序列(DNA或RNA)切割成一定长度,并连接上适配体,以便于后续测序。
而序列生成则是指将文库中的DNA或RNA片段高通量排列并进行测序,一般采用Illumina、PacBio等平台。
数据分析则是根据得到的序列数据进行比对、注释、变异分析等,可以使用相应的软件如Bowtie、BWA、SnpEff 等。
二、高通量测序技术的应用高通量测序技术的应用领域非常广泛,下面就对其中一些典型应用进行介绍。
1. 基因组学研究高通量测序技术的出现,让基因组学的研究有了巨大的进步。
利用高通量测序技术可以大规模的测序,通过数据分析建立新的物种数据库、基因注释、基因序列比较等工作。
例如常用的模式生物如小鼠、果蝇等,它们的基因组特性已经非常完善,并且注解、系统分析等软件也很成熟,但是对于许多生物资源的基因组测序比较缺乏,因此,高通量测序技术为这些生物测序提供了非常重要的工具。
2. 基因变异检测基因变异是指在DNA序列中出现的不同于人类参考基因组序列的突变或异型。
基因变异能引起遗传性疾病的发生或某些代谢物的降解速度的改变,进而影响个体的生命过程。
高通量测序技术可以实现测序数据的长读取长度和高的质量,为基因变异检测提供了强有力的工具。
这种技术可以将多个样本进行比对,找出共有的SNP,并计算影响SNP功能的染色体和环境条件等,进一步来实现对基因变异、基因突变等的检测。
3. 表观基因组学研究表观遗传学指代因表观遗传现象(如DNA甲基化、组蛋白修饰)弥补了经典遗传学无法解释某些遗传现象的缺口。
314芯片测序方法文库构建:PCR扩增基因组DNA 的目标区域1.选择下方合适的表格是基于用户选择的是2x的引物池还是5x的引物池。
对每一个DNA和引物池的混合,将以下组分加入PCR 管内。
以下表格提供了包含和未包含Ion AmpliSeq?Sample ID Panel的反应体系。
对多个反应可准备配置混合溶液。
2.充分震荡,简单离心。
3.将PCR管置于PCR仪内,运行一下程序以扩增基因组上目标区域。
*4分钟是对≤1536对引物池;8分钟是对1537-6144对引物池;16分钟是对6145-24576对引物池。
+PCR产物可置于10℃过夜储存。
注意:Ready‐to‐use panels: 当加入Ion AmpliSeq? Sample ID panel时不需要调整循环数。
Custom primer pools:如果一个custom 设计的panel中包含两个引物池,并分别落在不同的循环数分类中,请选用更大的循环数来扩增这两个引物池。
一般标准样品可以增加一个循环,从而保证其扩增效率,石蜡切片样品可增加2-3个循环。
如果引物池有两管及以上,应当分别扩增(10μl体系)。
扩增完后混合取20μl进行下轮实验。
暂停点– PCR产物可以储存在10°C过夜,需要更长时间储存,可以置于–20°C.部分消化引物序列1.轻度离心PCR管将反应后溶液收集到管底。
往每个混合反应液中加入2微升FuPa?Reagent?,总体系达到22微升。
2.充分振荡混匀,轻度离心将液体收集到管底。
(也可以选择用枪吸取一半以上的液体上下吹打至少5次来混匀)3.将PCR管放入PCR仪,按以下程序运行。
将接头连接至扩增产物并纯化配置并运行连接反应1.如果有可见的沉淀物在Switch溶液中,通过室温下震荡或上下吹吸来溶解并重悬。
2.小心PCR管加入以下成分到每个反应孔内消化样本。
在加入之前可先混合Switch溶液和接头。
3.在每个PCR管内加入2微升DNA连接酶。
4.充分振荡混匀,轻度离心将液体收集到管底。
(也可以选择用枪吸取一半以上的液体上下吹打至少5次来混匀)可以置于–20°C纯化未扩增的文库使用Agencourt? AMPure? XP试剂1.5x样本体积重要事项!–将AMPure??XP?reagent?置于室温并充分振荡将磁珠混匀,使用时缓慢吸取.接下来的步骤需要使用新鲜配置的70%乙醇。
对每个样品,混合230微升无水乙醇和100微升无核酶水。
1.将加完接头的文库转移至1.5ml离心管内,加入45微升(1.5x样本体积)的Agencourt?AMPure? XP试剂。
用枪吹打5次使DNA与磁珠悬浮液充分混匀。
2.将混合液置于室温5分钟。
3.将1.5ml离心管置于DynaMag?‐96?Side?Magnet?(Cat.?no.?12331D)磁力架上,静置2分钟或者等溶液变清澈。
小心地吸走并弃掉上清,不要扰动磁珠。
4.向离心管中加入150 μL新鲜配制的70%乙醇,在磁力上来回移动1.5ml离心管来洗涤磁珠,然后小心地弃去上清,不要扰动磁珠。
5.重复第4步,进行第二次洗涤。
6.确保乙醇液滴已全部从孔中吸走。
将1.5ml离心管放置于磁力架上,室温空气干燥5分钟,注意不要过度干燥。
7.将1.5ml离心管从磁力架上拿开,在每管中加入50 μL Low TE充分浸润磁珠。
充分振荡混匀,轻度离心将液体收集到管底。
(也可以选择用枪吸取一半以上的液体上下吹打至少5次来混匀)8.将1.5ml离心管置于磁力架上2分钟。
上清液中含有文库。
文库扩增:1.将1.5ml EP管从磁力架上取下,加50 μl Platinum?? PCR?SuperMix?High?Fidelity 和2?μl of?Library?Amplification?Primer?Mix。
Platinum? PCR SuperMix High Fidelity和Library Amplification Primer Mix需要事先混合好。
充分漩涡震荡后简单离心,或者用枪上下吹吸5次。
2.将1.5ml EP管放在磁力架上至少2分钟,将上清约50μl转移到一个干净的200μL PCR 内。
3.运行一下PCR程序纯化扩增后的文库第一轮纯化1.加入25μl (0.5X的样品体积)?Agencourt??AMPure??XP?Reagent到每个样品管中,用移液枪吹吸5次。
2.室温下静置5分钟。
3.将样品管放在磁力架上至少5分钟,直至溶液变澄清。
4.小心的将上清移入另一个干净的PCR管中。
第二轮纯化1.在第一轮纯化取到的上清中加入60μl Agencourt??AMPure??XP?Reagent,用枪头反复吹吸5次2.室温下静置5分钟。
3.将样品管放在磁力架上至少3分钟,直至溶液变澄清,将上清吸除。
4.加入150μl新鲜配置的70%的酒精,缓慢旋转两圈,将上清吸除。
5.重复步骤46.确保酒精完全被移除,打开管盖,常温干燥5分钟。
7.将样品管从磁力架上取下,加入50μl?Low TE,充分漩涡震荡,简单离心,或将移液枪调至40μl?吹吸5次。
8.将样品管放入磁力架后,静置两分钟,将上清转移到新的1.5ml离心管。
Qubit定量1.制备?Qubit??dsDNA?HS?reagent稀释液:1μl Qubit??dsDNA?HS?reagent加199μl?Qubit??dsDNA?HS?Buffer。
2.在10 μL?的?amplified?Ion?AmpliSeq??library?中加入190 μL?的dye?reagent,混合充分后静置2分钟。
3.1μL样品中加入199μL dye reagent进行测样。
定量完成后,不同引物的相同样品的管合为一管(相同浓度)每管定到300PM,混合后能达到100PM。
Ion PGM 200bp自动模本制备在Ion OneTouch 2 instrument上制备模板ISP1.4.1 Ion OneTouch 2仪器准备1.4.1.1 打开Ion OneTouch 2背面的电源开关;按触摸屏上OPEN LID,等待离心机盖打开;1.4.1.2参照图1将Recovery Tubes(Ion PGM TM Template OT2 Supplies 200)放置于IonOneTouch 2离心收集器的转子中,随后将Recovery Routers(Ion PGM TM Template OT2 Supplies 200)放置于相应位置,手动盖上离心机盖;图11.4.1.3 参照图2顺序将Ion OneTouch? 2 Amplification Plate(Ion PGM TM Template OT2Supplies 200)放置于加热模块中,将加热模块手柄压向自己身体方向,盖上IonOneTouch? Lid,导管固定于上端的卡口和正面右上角的支架上,将AmplificationPlate的注射针垂直插入Ion OneTouch? DL Injector Hub,直至接触到底部的Recovery Router,然后松开注射针1.4.1.4 Ion OneTouch? Oil和Ion PGM OT2 Recovery Solution添加1.4.1.4.1每次开始使用新的Ion PGM? Template OT2 200 Kit,丢弃掉前一个试剂盒使用的Ion OneTouch? Sipper和Ion OneTouch? Tubes,戴上新的乳胶手套,装上新的IonOneTouch? Sipper;将Ion OneTouch? Oil 瓶(Ion PGM TM Template OT2 Solutions200)上下颠倒10次,倒入任意一个新的Ion OneTouch? Tube 中,约1/2满,安装至标示有的下方相应位置拧紧;Ion PGM OT2 Recovery Solution(Ion PGM TMTemplate OT2 Solutions 200)倒入另一个新的Ion OneTouch? Tube 中,约1/3满,安装至标示有的下方相应位置拧紧;1.4.1.4.2 同一个Ion PGM? Template OT2 200 Kit的后续使用准备时,将Ion OneTouch? Oil(Ion PGM TM Template OT2 Solutions 200)瓶上下颠倒10次,取下有Ion OneTouch?Oil的Ion OneTouch? Tube,剧烈混匀至产生微小的气泡(图3),加入Ion OneTouch?Oil,约1/2满,安装至标示有的下方相应位置拧紧;将Ion PGM OT2 RecoverySolution(Ion PGM TM Template OT2 Solutions 200)倒入另一个的含有Ion PGM OT2Recovery Solution的Ion OneTouch? Tube 中,约1/3满,安装至标示有的下方相应位置拧紧;图31.4.2 Ion OneTouch反应液相准备及上机1.4.2.1 按照如下表格顺序室温配置体系,试剂来自Ion PGM TM Template OT2 Reagents 2001.4.2.2 将上述体系涡旋震荡5s,稍离心;1.4.2.3 将Ion PGM? Template OT2 200 Ion Sphere? Particles最大速漩涡混匀1min,上下吸打5次上下吸打混匀,室温放置备用;1Ion PGM OneTouch? Plus Reaction Filter(Ion PGM TM Template OT2 Reactions 200移液器将所有的液相缓慢从距离另外两个孔较远的样品孔(图4)注入reactionfilter,注意移液头与样品孔紧密接触插紧;图41.4.2.5 用1000ul移液器从此孔缓慢加入1000ul Ion OneTouch? Reaction Oil(Ion PGM TMTemplate OT2 Solutions 200),更换一个新的1000ul Universal Fit Filter Tip移液头(防止交叉污染),再缓慢加入500ul Ion OneTouch? Reaction Oil;1.4.2.6 如图5,将加样孔置于左侧,顺时针倒转reaction filter,避免管内导管接触反应液相;图51.4.2.7 如图6,将reaction filter插入Ion OneTouch?相应位置,边缘凸起位置朝向自己;凸起部分图61图71.4.2.9 选择Ion PGM TM Template OT2 200 kit(图8)图81xpert,最后点击NEXT开始运行。