蛋白质二级结构预测精编版
- 格式:ppt
- 大小:1.20 MB
- 文档页数:6

蛋白质二级结构的预测方法初探【摘要】提出了研究蛋白质二级结构预测的意义,介绍近三十多年来蛋白质二级结构预测的方法分类,分别列举出各类典型蛋白质二级结构预测方法的具体实现过程并最终对预测结果进行比较。
【关键词】蛋白质二级结构多序列比对法神经网络蛋白质的二级结构指多肽链本身通过氢键沿一定方向盘绕、折叠而形成的构象。
蛋白质分子并非如一级结构那样是展开的“线状”,而是出于更高级的水平,多肽链主链中各原子在各局部的空间排布如何,是蛋白质二级结构主要研究的问题。
蛋白质的功能主要由特定的三维结构所决定,因此,为了了解蛋白质功能,人们迫切需要确定蛋白质的三维结构。
目前测定蛋白质结构的方法有x-光线衍射、核磁共振以及电子显微镜方法。
所有这些方法都是耗时的,并且受到较多限制,如需纯净蛋白、小蛋白等。
这样结构测定技术远不能赶上每天数以千计的测序速度。
为了缩小结构与已知序列之间的差距,发展理论的蛋白质结构预测方法势在必行。
因此,在认为蛋白质的三维结构式由它的序列和环境所决定的情况下,促使人们利用蛋白质二级结构来预测其三维结构。
蛋白质二级结构预测问题已成为生物信息学的经典问题之一。
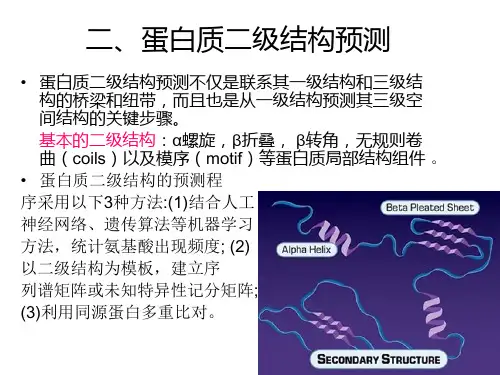
蛋白质二级结构预测已经有三十多年的历史,各种不同的预测方法可以分为三类:统计学方法、多重序列比对法、神经网络方法。
本文将例举三种典型性的预测方法进行阐述和比较。
chou—fasman是一种典型的统计学预测方法,基于15个已知构象的蛋白质和2473个氨基酸确定蛋白质二级结构。
它的经验规则是使用进行二级结构预测:寻找折叠核:从6个残基中找到了4个(hb或hb)便可以确定一个b折叠形成核,相反当(bb或bb)出现概率大于1/3时便不能确定;沿着多肽链向两个方向延伸b折叠形成核,直到遇到连续几个b折叠破坏者时才终止。
b折叠破坏者包括b4,b3i等等;边界调整:glu很少出现在b区,pro也不会出现在b折叠中,带点荷氨基酸残基都很少出现在两端。
trp频繁出现在n-末端。

《蛋白质的β-发夹、β(γ)-转角及四类简单超二级结构预测》篇一一、引言蛋白质是生命体系中最为基础和重要的组成部分之一,它们承担着众多生命活动所需的特定功能。
这些蛋白质分子是由线性序列的氨基酸组成,并折叠形成特定结构的复杂有机化合物。
在这篇文章中,我们将讨论三种关键的二级结构,即β-发夹、β(γ)-转角以及四种常见的超二级结构。
通过对这些结构的分析预测,我们能够更深入地理解蛋白质的结构与功能的关系。
二、β-发夹结构预测β-发夹结构是蛋白质中一种常见的二级结构,它由一系列连续的β-折叠片段组成,其中两个或多个β-折叠片段通过一个或多个弯曲的肽链连接起来。
这种结构在蛋白质中起到稳定和支撑的作用。
预测β-发夹结构通常需要利用生物信息学软件和算法,通过分析氨基酸序列的物理化学性质以及与其他已知结构的比对来完成。
三、β(γ)-转角结构预测β(γ)-转角是蛋白质中的一种弯曲结构,通常由数个氨基酸残基组成。
这种结构在蛋白质的折叠和功能中起着关键作用,它连接了不同的二级结构单元,使蛋白质能够形成复杂的空间结构。
预测β(γ)-转角结构需要分析氨基酸序列中的局部性质,以及结合其他二级结构和超二级结构的上下文信息。
这通常可以通过多种生物信息学软件和算法来实现。
四、四类简单超二级结构预测超二级结构是蛋白质中由若干个二级结构单元组合而成的更复杂的结构。
常见的四类简单超二级结构包括α螺旋束、β折叠片、无规则卷曲和螺旋-转角-折叠组合。
预测这些超二级结构需要综合考虑氨基酸序列的物理化学性质、二级结构的排列顺序以及与其他已知超二级结构的比对信息。
这通常需要借助生物信息学软件和算法进行大规模的计算和分析。
五、结论通过对蛋白质的β-发夹、β(γ)-转角以及四类简单超二级结构的预测,我们可以更深入地理解蛋白质的结构与功能的关系。
这些预测不仅有助于我们了解蛋白质在生命体系中的具体作用,还有助于我们设计和优化新的蛋白质结构,以实现特定的生物医学应用。

《蛋白质的β-发夹、β(γ)-转角及四类简单超二级结构预测》篇一一、引言蛋白质是生命体系中的基本组成部分,其结构决定了其功能。
在蛋白质的众多结构中,β-发夹、β(γ)-转角以及超二级结构等都是其重要的结构特征。
本文将针对这些结构进行预测分析,以期为蛋白质的结构与功能研究提供一定的理论基础。
二、β-发夹结构预测β-发夹结构是蛋白质中常见的一种二级结构,它由平行的β-折叠构成,并通过氢键等相互作用形成环状结构。
在预测β-发夹结构时,我们需要首先识别出连续的β-折叠片段,并判断其是否存在环状结构。
通常可以通过生物信息学软件和算法对蛋白质序列进行分析,以预测可能的β-发夹结构。
三、β(γ)-转角结构预测β(γ)-转角是蛋白质中连接两个或多个二级结构的结构单元,其具有独特的弯曲和转折特性。
在预测β(γ)-转角结构时,我们主要关注蛋白质序列中的弯曲区域,分析其弯曲程度和角度变化,从而判断是否存在转角结构。
这同样可以通过生物信息学软件和算法来完成。
四、超二级结构预测超二级结构是蛋白质中由多个二级结构单元组合而成的更高级的结构形式。
常见的四类简单超二级结构包括α-螺旋簇、β-发夹簇、α+β簇以及无规则卷曲簇等。
在预测超二级结构时,我们需要综合考虑蛋白质序列中的各种二级结构单元的组合方式和空间排列,通过算法分析得出可能的超二级结构类型。
五、方法与技术在进行蛋白质的β-发夹、β(γ)-转角及超二级结构预测时,我们主要依靠生物信息学软件和算法。
这些软件和算法可以通过分析蛋白质序列中的氨基酸组成、二面角等信息,预测出可能的二级结构和超二级结构。
同时,我们还需要结合蛋白质的三维结构信息,对预测结果进行验证和修正。
六、结论通过对蛋白质的β-发夹、β(γ)-转角及超二级结构的预测,我们可以更深入地了解蛋白质的结构特征,从而为其功能研究提供重要的理论依据。
然而,由于蛋白质结构的复杂性和多样性,预测结果仍需结合实验数据进行验证和修正。

蛋白质结构预测Protein Structure PredictionHaibo SunDepartment of BioinformaticsMininGene BiotechnologyG h lMarch 22, 2007背景结构分类:z一级结构也就是组成蛋白质的氨基酸序列z二级结构即骨架原子间的相互作用形成的局部结构,比如alpha螺旋,beta片层和loop区等l h b t lz三级结构即二级结构在更大范围内的堆积形成的空间结构z四级结构主要描述不同亚基之间的相互作用。
结构测定的实验方法z核磁共振z X光晶体衍射两种。
一级结构级结构预测基础预测基础:z 实验:在体外无任何其他物质存在的条件下,使得蛋白质去折叠,然后复性,蛋白质将立刻重新折叠回原来的空间结构z 物理学的角度讲,系统的稳定状态通常是能量最小的状态二级结构反向β-折叠α-螺旋β-转角三级结构Turn or coilAlpha-helix Beta-sheetLoop and Turn蛋白质结构预测•Sequence secondary structure 3D structure Sequence →secondary structure →3D structure →functionProtein Structure PredictionProtein Structure Prediction •Prediction is possible because–Sequence information uniquely determines 3D structure–Sequence similarity (>50%) tends to imply structuralsimilarity•Prediction is necessary because–DNA sequence data »protein sequence data »structuredata199419972002.102007.3 Sequence (Swiss Port)40,00068,000114,033261,513 Sequence(Swiss-Port)4000068000114033261513 Structure (PDB)4,0457,00018,83842,474Methods预测方法Comparative (homology) modeling (同源建模法) Construct 3D model from alignment to proteinithsequences withknown structureg(g)(折识别法)Threading (fold recognition) (折叠识别法Pick best fit to sequences of known 2D / 3D structures (folds)Ab initio / de novo methods (从头预测法)Ab initio/de novo methods(Methods(1)同源性(Homology)方法:理论依据:如果两个蛋白质的序列比较相似,则其结构理论依据如果两个蛋白质的序列比较相似则其结构也有很大可能比较相似。

ncbi蛋白质序列的二级结构
NCBI(National Center for Biotechnology Information)是一个提供生物医学和基因组学信息的数据库,它包含了大量的蛋白质序列数据。
蛋白质的二级结构是指蛋白质分子中由氨基酸残基之间的氢键和其他非共价键形成的空间结构。
NCBI数据库中的蛋白质序列可以通过一些工具和算法来预测其二级结构。
一种常用的预测蛋白质二级结构的方法是利用基于序列的预测算法,例如PSIPRED和JPred。
这些算法使用蛋白质序列的氨基酸组成来预测其可能的二级结构,包括α-螺旋、β-折叠和无规卷曲等。
这些预测结果可以在NCBI数据库中的相关蛋白质条目中找到。
另一种方法是利用实验技术,如X射线晶体学和核磁共振等,来直接解析蛋白质的二级结构。
这些实验技术可以提供更准确和直接的二级结构信息,但需要耗费大量时间和资源。
总的来说,NCBI数据库中的蛋白质序列可以通过预测算法和实验技术来研究其二级结构。
这些信息对于理解蛋白质的功能和结构具有重要意义,有助于生物医学和基因组学领域的研究和应用。



随着蛋白质结构数据的积累,人们开始注意到一些较简单的序列与结构关系。
可以利用各种氨基酸的疏水值定位蛋白质的疏水区域,通过疏水氨基酸出现的周期性预测蛋白质的二级结构。
Lim等人很早就对α螺旋和β折叠归纳出了一套预测模式。
例如α螺旋的轮状结构特征,轮的一侧通常处于蛋白质的疏水核心,另一侧则常处于亲水表面,如图7.2所示。
因此,α螺旋中亲疏水氨基酸残基的出现位置也就有一定的规律性,亲水残基多出现在亲水侧面,而疏水残基则多出现在疏水侧面,反映在序列上就是一些特征的亲疏水残基间隔模式。
疏水性氨基酸的位置有助于推断蛋白质中二级结构的定位,通过显示疏水氨基酸的分布分析二级结构。
例如,图7.2 是利用HELICALWHEEL程序画出的蛋白质蜂毒素旋轮图。
图中各个氨基酸沿螺旋排布,相邻氨基酸之间的旋转角度为100o。
疏水性氨基酸L、I和V位于螺旋的一侧,而亲水性氨基酸则分布在另外一侧,显示这个螺旋的两亲特性。
根据蛋白质序列中疏水性氨基酸出现模式,可以预测局部的二级结构。
例如,当我们在一段序列中发现第i、i+3、i+4位是疏水氨基酸时,这一片段就被可以预测为α螺旋;当我们发现第i、i+1、i+4位为疏水氨基酸时,这一片段也可以被预测为α螺旋。
同样,对于β折叠,也存在着一些特征的亲疏水残基间隔模式,埋藏的β折叠通常由连续的疏水残基组成,一侧暴露的β折叠则通常具有亲水-疏水的两残基重复模式。
不过,由于β折叠受结构环境的影响较大,序列的亲疏水模式不及α螺旋有规则。
原则上,通过在序列中搜寻特殊的亲疏水残基间隔模式,就可以预测α螺旋和β折叠。
在Biou等人提出的点模式方法中,将20种氨基酸残基分为亲水和疏水残基,用八残基片段表征亲疏水间隔模式。
以一个二进制位代表一个残基,疏水为1,亲水为0,共八位。
这样,八残基片段的亲疏水模式就可用1个0~255的数值来表示。
α螺旋的特征模式对应的值为9,12,13,17,……,201,205,217,219,237;β折叠的特征模式则由连续的1或交替的01构成。

了解点冷知识,蛋白质二级结构预测上一次小师弟给大家介绍了从蛋白质一级结构预测相关信息的网站——也即ExPASy ProParam的使用,今天我再给大家介绍一下蛋白质二级结构的预测。
在介绍具体的网站和软件使用之前,我想先介绍一下蛋白质二级结构预测的基础知识。
蛋白质二级结构有如下几种模式:α螺旋(当然,也存在其他形式的螺旋),β折叠,转角(turn),以及无规则卷曲(random coil)。
其中,无规则卷曲并不是一类真正的二级结构,只是作为一种分类,用来表示此类结构没有规则的二级结构。
需要说明的是,无规则卷曲也不是完全随机(random)折叠,其氨基酸分布以及折叠还是有一定规律的,只不过他们的结构非常灵活易变(flexible)。
甚至有研究表明,某些无规则卷曲是有明确而稳定的结构的。
关于二级结构的分类,更详细的可以参考DSSP(Dictionary of Protein Secondary Structure)分类系统,一共将蛋白质二级结构分为8类,其分类依据是二级结构中的氢键结合模式。
Loop,作为另一个在蛋白质二级结构层面的概念,并不是单指上述某一个类型的二级结构,而是一类多样化的二级结构,可以包括转角、无规则卷曲(long loops也被称作无规则卷曲)以及其它连接二级结构的氨基酸链。
Loops多数位于蛋白质分子表面,含有较多亲水氨基酸,具有灵活的构象,可以作为蛋白质的结合位点(比如抗体的抗原结合位点就由6个loop组成)以及酶的催化位点。
二级结构图片在蛋白质二级结构和三节结构之间,还存在两个概念,超二级结构和结构域。
超二级结构在很多教科书中也称作mofit(structural motif: 结构模体/结构基序,或者folding motif:折叠花式)。
超二级结构是指相邻二级结构在三维折叠中相互靠近所形成的组合,分为简单超二级结构和复杂超二级结构。
简单超二级结构一般只包含3个及3个以下的二级结构,我们熟悉的αα,ββ,以及βαβ结构模体就属于简单超二级结构,锌指结构(αββ)也是一种简单的超二级结构。