葡萄糖-磷酸脱氢酶缺乏症
- 格式:ppt
- 大小:2.02 MB
- 文档页数:5

疾病名:葡萄糖-6-磷酸脱氢酶缺乏症英文名:glucose-6-phosphate dehydrogenase deficiency缩写:别名:6-磷酸葡萄糖脱氢酶缺乏症;G-6-PD 缺乏症疾病代码:ICD:D55.0概述:红细胞葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G-6-PD)是红细胞糖代谢磷酸己糖旁路中的一个关键酶。
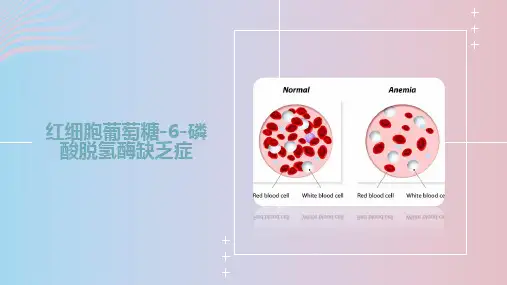
红细胞 G-6-PD 缺乏症(erythrocyte G-6-PD deficiency)是指红细胞G-6-PD 活性降低和(或)酶性质改变,导致以溶血为主要表现的疾病。
临床主要表现为先天性非球形红细胞溶血性贫血、新生儿黄疸、蚕豆病、药物性溶血、感染诱发溶血等。
如只有红细胞 G-6-PD 活性降低和(或)性质改变,而无溶血等临床表现,则称为红细胞G-6-PD 缺乏或缺陷。
在我国,红细胞 G-6-PD 缺乏症是遗传性红细胞酶缺乏症中最常见的一种。
流行病学:本病呈世界性分布,目前全球有超过2 亿人罹患G-6-PD 缺乏症。
携带G-6-PD 缺乏基因者估计占世界人口的7%,每年约出生450 万G-6-PD 缺乏儿。
G-6-PD 缺乏症的发生率和基因频率有明显的种族和地理分布差异,我国处于高发区。
欧洲、亚洲和我国均有“南高北低”的分布特点。
根据遗传学原理,G-6-PD 缺乏症最好用基因频率。
国内于 1983 年组成的全国G-6-PD 普查协作组曾对 7 个省、市、自治区的 9 个民族 19025(男性)进行了基因频率调查,基本摸清了我国此病的流行病学特点,即:①基因频率为 0.0000~0.4483,最高的基因频率发现于海南一个苗族半隔离群;②分布呈“南高北低”的趋势,北方各省出现一些散发病例,但相当一部分患者的祖籍在南方;③同一民族不同地区的基因频率有明显差别,而同一地区不同民族间反而差异不大,发病率分布的不均一据认为与恶性疟的自然选择有关。

血液内科葡萄糖-6-磷酸脱氢酶缺乏症患者诊治规范葡萄糖-6-磷酸脱氢酶(G-6-PD)缺乏症指因G-6-PD缺乏所致的溶血性贫血。
临床上可分为无诱因的溶血性贫血、蚕豆病、药物诱发和感染诱发的溶血性贫血以及新生儿黄疸5种类型。
(一)诊断要点1.高铁血红蛋白还原试验由于G-6-PD缺乏,红细胞不能生成足够的还原型烟酰胺腺嘌呤二核苷酸磷酸(NAD-PH),试管中加入亚甲蓝时,高铁血红蛋白还原少于正常值(75%以上)。
本法简便,适用于过筛试验或群体普查,缺点是有假阳性。
2.荧光斑点试验NADPH在长波紫外线照射下能显示荧光。
G-6-PD缺乏的红细胞内NADPH少,所以荧光出现延迟。
G-6-PD正常者10min 内出现荧光。
可依此推测G-6-PD活性,试验操作方便,采血少,特异性也高。
3.硝基四氮唑蓝纸片法红色滤纸片被G-6-PD正常红细胞还原成紫蓝色,严重G-6-PD缺乏者滤纸仍为红色。
4.红细胞海因小体计数在所采血中先加入乙酰苯肼,37℃温育后做甲基紫活体染色。
G-6-PD缺乏的红细胞内可见海因小体,计数>5%有诊断意义。
5.G-6-PD活性测定最为可靠,是主要诊断依据。
但在溶血高峰期及恢复期,酶活性可以正常或接近正常。
Zinkham法的正常值为(12.1±2.09)U/gHb(37℃)。
(二)临床类型及治疗1.无诱因的溶血性贫血属于先天性非球形细胞性溶血性贫血的一种,G-6-PD可低至0,温育后红细胞渗透性脆性正常,温育后自体溶血试验阳性,无血红蛋白病,抗人球蛋白试验阴性。
输血及使用糖皮质激素可改善病情,切脾效果不理想,需慎重。
2.蚕豆病由于食蚕豆后引起的溶血性贫血,是广东、广西、湖南、江西等地农村常见的血液病。
系蚕豆中何种物质引起尚无定论。
患者并非每年食蚕豆均发病,发病程度与食蚕豆量并不成比例。
患者绝大多数为儿童,3岁以上发病者占70%左右。
男性显著多于女性。
起病急骤,均在食新鲜蚕豆数小时突然发病。

葡萄糖-6-磷酸脱氢酶缺乏症的遗传学研究概述葡萄糖-6-磷酸脱氢酶缺乏症(G6PD缺乏症)是一种常见的遗传性红细胞酶缺陷性贫血。
本文将探讨G6PD缺乏症的遗传学特征、致病机制以及临床表现,以便更好地理解和管理该遗传性疾病。
一、遗传学特征G6PD缺乏症是X连锁隐性遗传方式的一种遗传性疾病。
这意味着男性只需要一个受影响的基因就可以表现出该缺陷,而女性则需要两个受影响的基因才会表现出该缺陷。
因此,男性的患者比例较高。
二、致病机制G6PD是体内重要的代谢酶之一,在细胞内起着氧化还原反应催化剂的作用。
当体内存在G6PD活性降低或完全丧失时,红细胞无法抵御氧化应激,导致氧化应激损伤和溶血。
此外,某些药物、感染和食物摄入等因素也可触发病发。
三、临床表现G6PD缺乏症的临床表现通常与红细胞溶血有关。
在孟加拉国、地中海地区和非洲等高发地区,尤其是登革热高发地区,G6PD缺乏症更容易引起急性溶血危机。
急性溶血反应可以导致黄疸、贫血以及肝脾肿大等症状,严重时还可能危及生命。
四、遗传咨询和筛查由于G6PD缺乏症的遗传方式与表型变异性复杂,遗传咨询和筛查是预防该病的重要措施。
对那些家族中有G6PD缺乏症患者或携带者的人进行基因检测可帮助确定个人是否具有遗传风险,并进行相应的建议和干预。
五、治疗与管理目前对于G6PD缺乏症还没有特定的治疗方法。
在急性溶血危机发作时,积极处理黄疸、维持液体平衡和红细胞输注是主要的治疗手段。
此外,避免接触过敏源和提供足够的营养也对预防和管理G6PD缺乏症患者至关重要。
结论葡萄糖-6-磷酸脱氢酶缺乏症是一种常见的遗传性红细胞酶缺陷性贫血。
通过了解其遗传学特征、致病机制以及临床表现,可以更好地辨识该疾病并进行有效的管理。
遗传咨询与筛查对于预防该疾病具有重要意义,而在治疗与管理方面目前仍需要进一步的探索和改进。
未来,我们希望能通过深入的遗传学研究为G6PD缺乏症的诊断、预防和治疗提供更为准确和有效的方案。

G6PD缺乏症俗称蚕豆病,是遗传性葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症是最常见的一种遗传性酶缺乏病,俗称蚕豆病。
我国是本病的高发区之一,呈南高北低的分布特点,患病率为0.2-44.8%。
主要分布在长江以南各省,以海南、广东、广西、云南、贵州、四川等省为高。
G6PD缺乏症发病原因是由于G6PD基因突变,导致该酶活性降低,红细胞不能抵抗氧化损伤而遭受破坏,引起溶血性贫血。
G6PD缺乏症属X连锁不完全显性遗传,以前认为是X连锁隐性遗传,后来发现一些女性杂合子G6PD酶活性也有不同程度降低、甚至患病,表现为X连锁显性遗传方式。
酶缺乏的表现度不一,因此本病可引起女性杂合子发病,就不能说是传男不传女的遗传规律了。
一些女性杂合子酶活性可能正常。
男性患者多于女性。
根据G6PD缺乏症特有的遗传方式,儿子患病,一定是从母亲遗传得到;女儿患病,可能是从父方,也可能是从母方,还有可能是从父母双方遗传而来。
什么是不完全显性遗传通常显性遗传的特点是父母任一方的致病基因只要传给子女,都可导致发病。
有些携带显性遗传基因的个体可无临床表现,即外显率不是100%,这种情况就是不完全显性。
正如G6PD 缺乏症一样,因属于X连锁,群体中出现外显不全现象,故称X连锁不完全显性遗传。
1.禁食蚕豆或蚕豆加工品,避免在蚕豆开花、结果或收获季节去蚕豆地。
2.禁止食用的食品:珍珠末,金银花,川莲,牛黄,腊梅花,熊胆,保婴丹。
3.禁止使用含萘的臭丸放入衣柜驱虫。
4.禁止使用的药物:乙酰苯胺、美蓝、硝咪唑、呋喃旦叮、呋喃唑酮、呋喃西林、苯肼、伯氨喹啉、扑疟母星、戊胺喹、磺胺、乙酰磺胺、磺胺吡啶、噻唑酮、甲苯胺蓝、SMZ、TNT等5.慎用的药物:扑热息痛、非拉西丁、阿斯匹林、氨基比林、安替比林、安坦、维生素C、维生素K、氯霉素、链霉素、异烟肼、磺胺嘧啶、磺胺胍、磺胺异恶唑、氯喹、秋水仙碱、苯海拉明、左旋多巴、苯妥英钠、普鲁卡因酰胺、乙胺嘧啶、奎尼丁、奎宁、SM、TMP、优降糖等6.感染性诱因:病毒性肝炎、流感、肺炎、伤寒、腮腺炎等。

G6PD缺乏症(红细胞葡萄糖-6-磷酸脱氢酶缺乏症)G6PD缺乏症,是遗传性葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症,是最常见的一种遗传性酶缺乏病,俗称蚕豆病。
G6PD缺乏症发病原因是由于G6PD基因突变,导致该酶活性降低,红细胞不能抵抗氧化损伤而遭受破坏,引起溶血性贫血。
疾病简介红细胞葡萄糖-6-磷酸脱氢酶(G-6-PD)缺乏症是世界上最多见的红细胞酶病,据统计全球约有近4亿人G-6-PD缺陷。
本病常在疟疾高发区、地中海贫血和异常血蛋白病等流行地区出现,地中海沿岸、东南亚、印度、非洲和美洲黑人的发病率较高。
我国是本病的高发区之一,分布规律呈“南高北低”的态势,长江流域以南,尤以广东、海南、广西、云南、贵州、四川等地为高发区,发生率为4%-15%,个别地区高达40%。
中医学名G6PD缺乏症其他名称蚕豆病英文名称glucose-6-phoshate dehydrogenase deficiency;G-6-PD所属科室内科- 血液内科主要症状慢性溶血性贫血、急性起病、贫血、黄疸、血红蛋白尿主要病因进食新鲜蚕豆禁用磺胺嘧啶、SMZ、SMZ—TMP等传染性无传染性疾病分类本病有多种G-6-PD基因变异型,伯氨喹啉型药物性溶血性贫血或蚕豆病,感染诱发的溶血,新生儿黄疸等。
发病原因诱因有:①蚕豆;②氧化药物:解热镇痛药、磺胺药、硝基呋喃类、伯氨喹、维生素K、对氨基水杨酸等;③感染:病原体有细菌或病毒。
发病机制本病是由于调控G-6-PD的基因突变所致。
呈X连锁不完全显性遗传。
由于G-6-PD基因的突变,导致红细胞葡萄糖磷酸戊糖旁路代谢异常,当机体受到伯氨喹啉型药物等氧化物侵害时,氧化作用产生的H2O2不能被及时还原成水,过多的H2O2可致血红蛋白和膜蛋白均发生氧化损伤。
最终造成红细胞膜的氧化损伤和溶血。
溶血过程呈自限性,因新生的红细胞G-6-PD活性较高,对氧化剂药物有较强的“抵抗性”,当衰老红细胞酶活性过低而被破坏后,新生红细胞即代偿性增加,故不再发生溶血。

葡萄糖-6-磷酸脱氢酶缺乏症是怎么回事*导读:本文向您详细介绍葡萄糖-6-磷酸脱氢酶缺乏症的病理病因,葡萄糖-6-磷酸脱氢酶缺乏症主要是由什么原因引起的。
*一、葡萄糖-6-磷酸脱氢酶缺乏症病因1.G-6-PD及其生化变异型“正常”酶称之为G-6-PD B,G-6-PD缺乏症是由于编码G-6-PD氨基酸序列的G-6-PD结构基因异常所致。
部分纯化残存酶的详细的生化研究提示它们之间存在异质性,这些异常的酶即为G-6-PD生化变异型。
1966年,世界卫生组织(WHO)在日内瓦召开的国际会议上对G-6-PD变异型的命名、分型标准及方法作了统一规定。
G-6-PD的定型主要根据电泳速率及酶动力学特征参数,诸如酶活性、电泳速率、6-磷酸葡萄糖(G6P)和辅酶Ⅱ(NADP)的米氏常数(KM),底物同类物(去氧G6P、磷酸半乳糖、脱氨NADP、辅酶Ⅰ)利用率、热稳定性、最适pH,但最低限度需要下列5项:①酶活性;②电泳速度;③G-6-PD米氏常数;④去氧G6P的相对利甩率;⑤热稳定性。
目前,国际上现已报道的G-6-PD变异型有400余种,其中约300种是按WHO推荐的标准方法进行鉴定的,还有大约100种变异型是采用其他方法鉴定的。
根据这些变异型的酶活性和临床意义分为5大类:第1类变异型活性非常低(低于正常的10%)伴有终身溶血性贫血;第2类变异型,尽管体外活性非常低,但不伴有慢性溶血,只有在某些特殊情况下才会发生溶血,这1类型是常见的类型如G-6-PD地中海(Mediterranean)型;第3类变异型其酶活性为正常的10%~60%,只有在服用某些药物或感染时才会发生溶血;第4类变异型是由于不改变酶的功能活性的突变所致;第5类变异型其酶的活性是增高的。
第4和第5类没有临床意义。
在中国人中已在香港、台湾和海外华侨中发现12种,杜传书等在广东、海南、贵州、四川、贵阳、云南等省发现35种,其中12种为世界上的新类型。
国人变异型主要属于第2和第3类变异型。

g6pd的组成
葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症俗称蚕豆病,是最常见的一种遗传性红细胞缺陷性疾病,属于基因缺陷,由父母遗传给孩子。
G6PD属于人体代谢系统的重要组成物质,对吸收代谢起到比较重要的作用。
G6PD由G6PD基因编码,定位于X染色体xq28高密度区,长度18kb,由13个外显子和12个内含子组成。
G6PD有两种细胞异构体,活性形式为二聚体或四聚体。
G6PD缺乏患者在食用蚕豆、蚕豆制品或接触蚕豆花粉后引发红细胞破裂,血红蛋白经过尿液被排出体外,导致重度贫血,尿出酱油尿,精神不济,如果不及时治疗,可致昏迷、惊厥和急性肾衰竭,孩子甚至有生命危险,所以发病后应及时就医诊治。

葡萄糖-6-磷酸脱氢酶缺乏症[概述]葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症的主要特点是氧化性损伤、自限性溶血、诱因和临床表现不均一。
G6PD可催化生成辅酶II(NADPH),用以维持GSH还原状态,避免红细胞受氧化物的损害。
含氧化性基团的药物(如多种解热镇痛剂)和食物(如蚕豆中的蚕豆嘧啶)、细菌和病毒性感染、机体应激状态突然变化等因素,均可导致G6PD缺乏的红细胞溶血。
该病地理分布广泛,种族间发病率差异很大,我国的发病率在4%-15%,华南、西南地区为高发区。
G6PD缺乏作为遗传背景,根据不同的诱因,在临床上主要有5种表现类型,即新生儿黄疸、先天性非球形红细胞溶血性贫血(GNSHA)、蚕豆病、药物性溶血和感染性溶血。
[临床表现]1.急性溶血和慢性溶血均可见,以溶血急性发作多见。
2.任何年龄均可发生,以婴幼儿多见,男性显著多为女性。
3.年轻型患者无溶血发作时可无明显贫血、黄疸,脾脏轻度肿大或无肿大。
4.急性溶血主要表现为血管内溶血,贫血、黄疸伴有酱油色血红蛋白尿。
重者可出现溶血危象。
极重型患者可出现休克、肾衰竭。
5.G6PD缺乏症所致急性溶血的特点是具有自限性,即当溶血达到一定程度时,引起溶血的诱因虽未解除,溶血过程不再发展,恢复过程长短与患者酶缺乏程度有关。
[诊断要点]1.病史、溶血诱因及籍贯(1)有新生儿黄疸史、CNSHA和(或)急性溶血史。
(2)近期(1~7天内)食用蚕豆或蚕豆制品;母亲食用蚕豆后,患儿吸吮其母乳而发病。
(3)近期患病毒或细菌性感染。
(4)近期食用药物后出现溶血症状。
(5)患者籍贯多见于华南、西南地区,其他地区散在发生。
2.临床症状(1)慢性期:具有轻度慢性溶血性贫血指征,脾无明显肿大。
(2)发作期:具有急性血管内溶血指征,血红蛋白和红细胞计数急剧下降,皮肤巩膜黄染,尿液呈酱油色或浓茶色,可伴有畏寒、发热、呕吐、腰腹疼痛等。
(3)家系中男性患者症状明显严重。
3.实验室数据(1)非特异性溶血指标:1)符合溶血指征,尤其是血管内溶血指征如血红蛋白尿检阳性。

葡萄糖6-磷酸脱氢酶缺乏症G-6-PD缺乏症,全称为红细胞葡萄糖6-磷酸脱氢酶缺乏症,俗称“蚕豆病”,是一种染色体异常的遗传性溶血性疾病。
约90%发生于男性,且多见于3岁以下的儿童,是新生儿黄疸的重要原因。
G-6-PD(红细胞葡萄糖6-磷酸脱氢酶)能协助葡萄糖进行新陈代谢,并可产生一种能够保护红细胞的物质,以对抗某些“不良”物质的破坏。
而当体内缺乏G-6-PD时,红细胞失去了保护物质,容易受到某些“不良”物质的破坏,从而发生溶血。
主要表现为溶血症状,出现脸色苍黄、疲累、食欲差、黄疸、茶色尿等。
严重时,可能会出现昏迷,甚至死亡。
目前,还没有任何药物能治愈此病。
G-6-PD缺乏症在无诱因不发病时,与正常人一样,无需特殊处理。
防治的关键在于预防,请严格遵照以下健康处方:1、禁食蚕豆及其有关制品(如蚕豆酥、怪味豆),避免在蚕豆开花、结果或收获季节去蚕豆地。
2、衣橱、厕所等处不可以使用樟脑丸(含萘的臭丸)。
3、不要使用龙胆紫(紫药水)。
4、禁止使用的药物:乙酰苯胺、美蓝、硝咪唑、呋喃旦叮、呋喃唑酮、呋喃西林、苯肼、伯氨喹啉、扑疟母星、戊胺喹、磺胺、乙酰磺胺、磺胺吡啶、噻唑酮、甲苯胺蓝、SMZ、TNT等。
5、慎用的药物:扑热息痛、非拉西丁、阿司匹林、氨基比林、安替比林、安坦、维生素C、维生素K、氯霉素、链霉素、异烟肼、磺胺嘧啶、磺胺胍、磺胺异恶唑、氯喹、秋水仙碱、苯海拉明、左旋多巴、苯妥英钠、普鲁卡因酰胺、乙胺嘧啶、奎尼丁、奎宁、SM、TMP、优降糖等。
6、注意下列感染性诱因:病毒性肝炎、流感、肺炎、伤寒、腮腺炎等。
7、凡感染后或接触/服用以上食物或药物数小时或数天内,出现发热、腹痛、呕吐、面黄或苍白、尿呈黄褐色或暗红色等症状,属急性溶血反应,应立即到医院急诊科就诊!。

g6pd缺乏症诊断标准G6PD缺乏症,全称葡萄糖-6-磷酸脱氢酶缺乏症,是一种遗传性疾病,主要限于男性。
本文将介绍G6PD缺乏症的诊断标准及相关指引。
一、临床表现G6PD缺乏症在临床上可表现为溶血性贫血,其程度多因个体之间的差异而各异。
患者会出现贫血、黄疸、脾肿大等症状。
此外,患者还可能出现发热、腹痛、血尿等退行性溶血危象。
二、实验室检查对于临床怀疑G6PD缺乏症的患者,可通过实验室检查来确定诊断。
1. G6PD活性检测:G6PD缺乏症的诊断主要依靠G6PD活性测定。
可采用离心法、比色法或荧光测定法来测定红细胞中G6PD酶活性的水平。
2. 贫血检测:进行全血细胞计数,并检查红细胞相关指标,如红细胞计数、血红蛋白、红细胞比容等。
3. 溶血指标:检测患者的间接胆红素、血清铁蛋白、尿胆原等指标,以评估溶血程度。
4. 红细胞形态学检查:观察患者的红细胞形态,如有不规则形状或红细胞片状出现时,提示溶血性贫血可能与G6PD缺乏症相关。
三、诊断标准目前,关于G6PD缺乏症的诊断,尚无统一的国际标准,各国或地区会根据当地的人群特点和病情表现制定诊断标准。
1. 临床症状与体征:患者出现有贫血、黄疸、脾肿大等症状或体征。
2. G6PD活性:G6PD活性测定显示低于正常范围,但要注意,有些患者在发作期间G6PD活性水平可能正常。
3. 家族史:患者家族中有G6PD缺乏症的病史。
4. 基因检测:通过基因检测,发现G6PD酶编码基因突变,可以进一步确认G6PD缺乏症的诊断。
综上所述,要诊断G6PD缺乏症,需要结合临床症状与体征、G6PD活性测定、家族史和基因检测等综合判断。
四、鉴别诊断对于患者的临床症状与实验室检查结果存在疑惑时,需要进行鉴别诊断,以排除其他引起溶血性贫血的原因。
常见的鉴别诊断包括遗传性球形红细胞增多症、自身免疫性溶血性贫血、感染性溶血性贫血等等。
五、治疗与管理针对G6PD缺乏症患者,治疗应采取综合措施。
在发作期间应注意休息、补充营养、预防感染等,同时避免接触引起溶血的药物和食物。
g6pd缺乏症确诊标准G6PD缺乏症是一种遗传性疾病,主要影响葡萄糖-6-磷酸脱氢酶(G6PD)的功能。
它是一种酶缺乏性疾病,当身体缺乏这种酶时,红细胞受到氧化应激时会发生溶血。
G6PD缺乏症的确诊通常涉及以下几个方面:1.临床症状:G6PD缺乏症在大多数情况下是无症状的,但在暴露于氧化药物或特定食物后,患者可能会出现溶血性贫血的症状。
这些症状包括黄疸、腹痛、尿液变色(发红或发蓝)和贫血。
如果患者出现这些症状,尤其是在暴露于氧化药物或特定食物后,医生可能会怀疑其患有G6PD缺乏症并进行进一步检查。
2.基因检测:G6PD缺乏症是由G6PD基因的突变引起的,因此进行基因检测可以确定G6PD缺乏症的诊断。
通过提取患者的DNA样本,可以进行突变的基因检测。
目前已知有数十种不同的突变与G6PD缺乏症相关,其中最常见的是G6PD A-、G6PD Mediterranean和G6PD Canton 等。
3.红细胞酶活性检测:红细胞酶活性检测是确诊G6PD缺乏症的主要方法。
该检测可以确定G6PD的活性水平,因为G6PD缺乏症患者的红细胞内G6PD活性通常降低或完全缺乏。
这种检测一般通过采集患者的血液样本进行实验室检查来完成。
在确诊G6PD缺乏症后,医生通常还会了解患者的病史和家族病史,以确定疾病的类型和严重程度。
此外,医生还可能要求患者进行进一步的检查,如血红蛋白测定、胆红素测定、骨髓穿刺等,以评估溶血性贫血的程度和其他可能存在的并发症。
需要指出的是,G6PD缺乏症的确诊需要综合考虑多个因素,特别是在没有症状的患者中。
在一些情况下,可能需要进行多次酶活性检测以确保准确性。
因此,确诊G6PD缺乏症需要深入的临床评估和实验室检查,最好由经验丰富的医生进行诊断。
总之,G6PD缺乏症的确诊主要依靠临床症状、基因检测和红细胞酶活性检测。
这些检查可以帮助医生确定患者是否患有G6PD缺乏症,并确定疾病的类型和严重程度,从而为对该疾病的管理和治疗提供指导。
葡萄糖-6-磷酸脱氢酶缺乏症,又名G6PD缺乏症(英文:G lucose-6-P hosphate D ehydrogenase deficiency),俗称蚕豆症,是一种常见的先天遗传性疾病。
患者由于遗传基因的先天缺陷,无法正常地分解葡萄糖。
除此以外,部份药物和化学物如蚕豆、樟脑、臭丸、龙胆紫(紫药水)、都会令患者出现急性溶血反应,症状包括黄疸、精神不佳,严重时会出现呼吸急速、心脏衰竭,甚至会出现休克而有生命危险。
由于先天性六磷酸葡萄糖去氢酵素缺乏症(以下简称为G6PD)是X染色体联锁遗传性疾病,而男性只得一条X-染色体,故此病症几乎只出现于男性身上,但带有此病因的女性亦有可能出现轻微的症状。
以下为G6PD发病时可能出现的症状:∙持续的黄疸∙溶血反应,由以下项目引发:∙某类药物(见下)∙某类食物(如蚕豆、金银花)∙其他物品(如樟脑、龙胆紫(紫药水))∙其他疾病(如受到严重病菌感染、糖尿病)∙严重症状可引致急性肾衰竭诱发G6PD症状的药物有:∙伯氨喹(Primaquine)∙奎宁、汤力水(tonic water)等抗疟药物∙磺胺类抗生素∙砜类:如用以治疗麻疯病的氨苯砜∙其他含硫磺的药品,如治疗糖尿病、控制血糖的药物血糖平(Glibenclamide)∙呋喃妥因:治疗尿道感染的抗生素∙阿司匹林当某些族裔的病人,出现黄疸、贫血,以及对某些诱因产生溶血反应时,又或是家族中有G6PD患者,都会被列为G6PD 的疑似个案,需要作进一步的测试。
G6PD的测试包括:∙全套血球计数(Complete blood count)及网状细胞(reticulocyte)计数;当症状出现时,G6PD患者的海因兹小体(Heinz bodies)会出现于在检验血液玻片的红血球内。
∙肝脏蛋白酶测试,以剔除其他黄疸症状的诱因∙结合珠蛋白(Haptoglobin)于溶血反应中会减少∙库氏试验(直接抗球蛋白试验,Coombs test):于G6PD患者病发时应呈阴性反应,这是由于G6PD引发的溶血反应并非由免疫系统协调产生∙甲状腺功能量度(TSH measurement)当有足够证据支持病者症状由G6PD引发,便可以"Beutler fluorescent spot test"为患者进行直接测试,其他可能的诊断方法,有DNA直接测试及检查G6PD的基因排列。
红细胞葡萄糖-6-磷酸脱氢酶缺乏症(glucose-6-phosphate dehydrogenase deficiency,简称G6PD缺乏症)是一种最常见的遗传性溶血性疾病,目前溶血机制尚不十分明确。
该病的临床表现与一般溶血性贫血大致相同,患者多数平时不发病,无症状。
发病时多见于蚕豆和某些药品、感染等引起的急性溶血性贫血,极少数患者则表现为慢性非球形细胞溶血性贫血。
该病是新生儿高胆红素血症的主要发病原因之一,严重者可致新生儿核黄疸,造成永久性的神经损害以致智力低下,甚至死亡。
世界范围内约有4亿人受累,世界卫生组织推荐G6PD缺乏症患病率超过5%的地区常规筛查G6PD活性。
研究显示该病呈世界性分布,主要在非洲热带、中东、亚洲热带和地中海某些地区、巴布亚新几内亚等地区高发。
我国呈现“南高北低”,以长江流域以南为主,北方发病率较低。
我国的相关筛查情况:四川省成都地区发病率约6.1%(2013);海南省发病率为1.46%(2013);广东省玉林市新生儿缺乏症阳性率为 5.85%(2010);山东省聊城地区阳性率为 1.6%(2012);广东省东莞市G6PD缺乏率为3.33%(2012);云南省昆明市发病率是0. 6%(2012);广东省顺德区发病率为4.04%(2013);贵州省发病率为3.43% (2001);云南省傣族的部分地区发病率高达20%(2007)。
既往的研究受检测技术所限,女性杂合子由于酶活性有较大的异质性,生化检测方法常常不能检出,影响了群体出生缺陷干预工作的实施。
外显子1372的1376M、1388M和外显子2的95M突变仅在华人中发现,是中国人最主要的基因突变类型,占突变的68. 5%~91.0%以上。
检测东南亚地区及中国人常见的(包括了80%以上的突变类型) 11种G6PD基因突变( 95A→G、392G→T、487G→A 、493A→G、592C→T、871G→A、1024C→T、1311C →T、1360C→T、1376G →T 、1388G→A )。
g6pd实验室检查项目摘要:1.概述G6PD缺乏症2.G6PD实验室检查项目的意义3.G6PD实验室检查方法及结果解读4.G6PD缺乏症的预防与治疗5.结论正文:**G6PD缺乏症概述**葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症是一种遗传性溶血性贫血病。
患者体内的G6PD酶活性降低,导致红细胞无法正常代谢,从而容易受到氧化应激损伤,引发溶血反应。
G6PD缺乏症在全球范围内都有较高的发病率,尤其在发展中国家。
我国人群中G6PD缺乏症的患病率约为4%-15%。
**G6PD实验室检查项目的意义**G6PD实验室检查项目旨在检测患者G6PD酶活性,协助诊断G6PD缺乏症。
通过检查,可以了解患者红细胞对氧化应激的敏感性,为临床确诊和分型提供依据。
此外,G6PD实验室检查还对评估病情、监测治疗效果和预测溶血风险具有重要意义。
**G6PD实验室检查方法及结果解读**G6PD实验室检查主要采用定性试验和定量试验两种方法。
定性试验包括磷酸定量法、硫酸铜法等,可以快速筛查G6PD缺乏症。
定量试验如荧光定量法,可以精确检测G6PD酶活性,为临床提供更为可靠的诊断依据。
检查结果解读时,需关注以下指标:1.正常值范围:不同方法的正常值范围略有差异,一般磷酸定量法为1.2-2.5U/gHb,硫酸铜法为0.6-1.2U/gHb。
2.活性降低:G6PD酶活性低于正常值,提示可能存在G6PD缺乏。
3.活性缺失:G6PD酶活性接近于零,确诊为G6PD缺乏症。
**G6PD缺乏症的预防与治疗**1.预防:避免接触氧化剂,如氧化性药物、化学制品等。
新生儿和儿童期尽量避免感染,以免加重溶血症状。
2.治疗:针对不同病情,采取相应治疗措施。
轻度缺乏者可观察治疗,中重度缺乏者需药物治疗,如糖皮质激素、雄激素等。
严重病例可考虑输血或造血干细胞移植。
**结论**G6PD实验室检查项目在诊断和治疗G6PD缺乏症中具有重要价值。