第六讲第一原理计算方法简介及MaterialsStudio中Castep使用方案
- 格式:ppt
- 大小:2.49 MB
- 文档页数:99





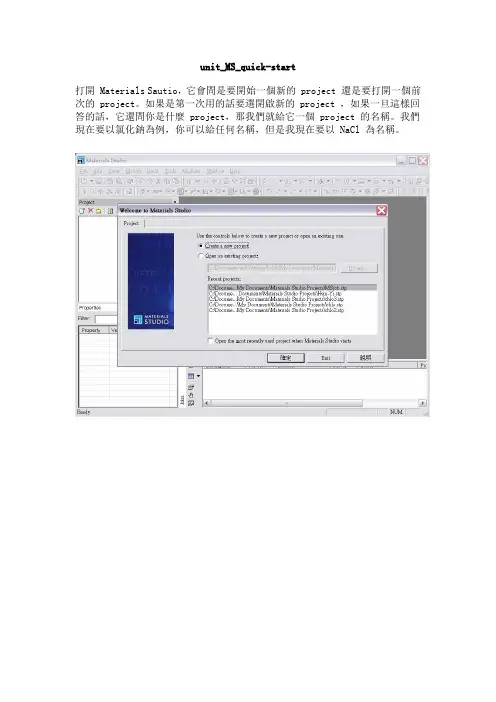
unit_MS_quick-start打開 Materials Sautio,它會問是要開始一個新的 project 還是要打開一個前次的 project。
如果是第一次用的話要選開啟新的 project ,如果一旦這樣回答的話,它還問你是什麼 project,那我們就給它一個 project 的名稱。
我們現在要以氯化鈉為例,你可以給任何名稱,但是我現在要以 NaCl 為名稱。
一開始進來要先介紹幾個重要的視窗,它們關係到我們進行模擬計算時所會處理及操作到的對象。
姑且可以分為這三類:一、進行計算的工作,己跑完的、正在跑的都算;二、計算工作總是有各有些不同的輸入與輸出檔案,我們經常會需要審視結果、修改輸出入的相關設定;三、材料的原子及電子結構 3D 模型帶有很多我們想要知道之關於這個材料的物性資料,例如晶體的晶胞邊長、原子的元素種類等等。
從 Veiw 的 Explorer ,它有三個 Explorer,job Exploroer、project exploroer、property explorer 。
job explorer 的開跟關是這樣按一次它就開起來。
這個是你跑什麼 job 近端遠端它都可以顯示,跑完了沒有、要不要把它移除等等,在這邊都可以操作,有很多 job 的時候會很好用。
project explorer 預設值是開著的,就是靠左邊垂直的這一塊,裡面對於跑 project 的相關物件,如文字輸出、3D結構等等都是在這裡選取,很像微軟視窗 (MS Windows) 裏頭的『檔案總管』。
要做東西總是需要選取一個 job 相關的目錄等等,所以 job explorer 在操作上來講是很重要的。
另外我也常常會打開的是 property explorer ,property explorer 在 MS 是新的東西,相對 Cerius2 而言是新的東西。
在 Cerius2 裡如果你想要知道一些 3D 物件的屬性,像是鍵長、鍵角,晶胞內原子數,就要分別去打開一些相關的表單,它才會印給你看,然而初學者還得學會這些表單藏在那裏。


信息记录材料2019年9月第20卷第9期(借息:技术与应用〕简述第一性原理计算软件CASTEP在材料物理教学中的应用吴玉辉(长春理工大学材料科学与工程学院吉林长春130022)【摘要】CASTEP程序是Cambridge Sequential Total Energy Package首字母的缩写,是一个基于第一性原理的量子力学程序.它是采用平面波贋势基组结合密度泛函理论,用来研究与设计材料物理性质功能强大的工具。
在教学过程中引入CASTEP计算模拟软件,对材料物理教学中的知识点及基本原理进行计算及演示,可以使教学内容和过程更加生动形象。
使与材料物理相关的知识点、更易于被学生掌握和接受,从而提高教学效率,激发学生兴趣。
本文旨在探索使用计算模拟软件在材料物理教学中应用,为材料物理的教学进行有益的探索及尝试。
【关键词】材料物理;CASTEP;Materials Studio;教学演示;第一性原理【中图分类号】TP37【文献标识码】A1简介第一性原理计算模拟软件CASTEP是Materials Studio软件包中的一个计算模块,它最初由剑桥大学-卡文迪什实验室的TCM(Theory of Condensed Matter group)凝聚态固体物理小组在20世纪80年代末、90年代初釆用Fortran77开发(后在2001年采用Fortran 95改写,用以提高整个代码的并行性和可持续性)。
CASTEP主要用于固体物理中凝固态相关性质的计算,20世纪90年代中期,由美国的分子模拟国际Molecular Simulations Inc.(MSI)公司发放许可证进而商业化运行,剑桥大学获得了一部分版税。
该公司后来与Genetics Computer Group(GCG)公司、英国的Synopsys Scient 迁ic系统公司以及Oxford Molecular Group(OMG)公司,于2001年6月1日合并组建了Accelrys公司,它是国际上知名的分子生物学及信息学公司,2016年1月被法国Dassault Systemes公司收购,随后Accelrys更名为BIOVIAo这个公司目前是全球范围内唯一能够提供分子模拟、材料设计以及化学信息学和生物信息学全面解决方案和相关服务的软件供应商。


计算机在材料科学与工程中的应用实验报告任课教师:学号:实验一 第一性原理计算1. 实验目的(1) 掌握第一性原理和密度泛涵的计算方法; (2) 学会使用Visualizer 的各种建模和可视化工具; (3) 熟悉CASTEP 模块的功能。
2. 实验原理CASTEP 是基于密度泛涵理论平面波赝势基础上的量子力学计算。
密度泛涵理论的基本思想是原子、分子和固体的基本物理性质可以用粒子密度函数进行描述。
可以归纳为两个基本定理:定理1:粒子数密度函数是一个决定系统基态物理性质的基本参量。
定理2:在粒子数不变的条件下能量对密度函数变分得到系统基态的能量。
不计自旋的全同费米子的哈密顿量为:H T U V =++其中动能项为:()()T dr r r ψψ+=∇∇⎰库仑作用项为:11'()(')()(')2'U drdr r r r r r r ψψψψ++=-⎰V 为对所有粒子均相同的局域势u(r)表示的外场影响:()()()V dru r r r ψψ+=⎰粒子数密度函数为:()()()r r r ρψψ+=ΦΦ对于给定的()r υ,能量泛函[]E ρ定义为:[]()()E dr r r T U ρυρ=++Φ⎰;[]F T U ρ=Φ+Φ系统基态的能量:'''''[]''''[][]()()[][]()()[]E T U V GE F dr r r E G G F dr r r E G ρρυρφρυρρΦ=Φ+Φ+ΦΦ==+>⎰=+=⎰3. 实验内容实验 1. 材料的电子结构计算;实验 2. 晶体材料的晶格[点阵]参数预报(要求材料体系为金属合金、化合物半导体或有机高分子材料);4. 实验设备和仪器(1) 硬件:多台PC 机和一台高性能计算服务器。
(2) 软件:主要利用Materials Studio 软件包里的Materials Visualizer 和CASTEP(Calculation 和Analysis)模块。
CASTEP 实战守则模型选定我们在进行材料物理模拟所需要做的第一步 (也是很重要的一步) 是模型的选定或建构。
CASTEP 虽然内建了很多功能来预测晶胞参数 (边长,夹角) 与原子位置,但仍然仰赖使用者告诉它 "要进行计算的系统是什么"。
在选定模型时,我们需切记如果系统内原子太多或是超晶胞体积太大,则计算量都会以平面波数的 3 次方增加到计算机难以负荷或使用者难以等待的程度,]此,我时时应考虑设计出一个足以表现出我们所想要研究的物理,而却又能使所有采用的超晶胞越少越好的模型。
如果研究上涉及到一系列原子数不同的大小系统需要做计算,最好能先计算小结构,不要一开始就送入大结构到计算机中。
如果要仿真的系统是含有杂质,则单位晶胞必须进一步放大成超晶胞以便使化学成分里的分数变成整数,因此晶胞会变得很大。
在某些特殊的情况,相互取代的元素种类是很类似的,(即在化学行为上类似),则下一个版本的CASTEP会提供一种叫做虚拟晶体近似 (Virtural Crystal Approximation,VCA) 的方法,则模型里面的原子就可以指定成如0.3A元素加0.7B元素这种样子,因此总可以以最小晶胞来做计算的模型。
但这种方法的精确度通常只适用于合金材料,故要小心使用。
模型的选定有许多人为抉择会含在里面,例如表面计算的层数,因此有些情况也需要进行所谓的收敛性测试。
Vps (poseudopotential)选择Vps选单:MS接口的设定是选用USP优先于NCP,USP有加速计算与减少内存使用的效果,其精确度也与NCP (norm-conserving potential) 相当。
至于什么时候使用NPC 呢 (就是在poseudopotential 选项中那些延伸文件名是 .recpot者),使用到NCP的场合有:1.某些CASTEP计算的功能尚未支持到USP,因此需要选用NCP。
2.为了要与已经发展的文献比较或进行验证3.对计算的结果存疑者,能提供『多一种选择』( 注:至于延伸档名是*.psp者也是属于norm-conserving的一种,是TM potential,在文献上也常被使用,但所需的截止动能较高,因此计算代价较大。
硅晶体能带结构的第一性原理计算班级:材料科学与工程3班学号:3015208064姓名:黄慧明一、实验目的通过实际操作初步的了解和掌握Materials Studio,基本掌握CASTEP 模块的操作步骤。
通过学习Materials Studio 软件,能够独立的进行简单的固体结构模型的构造和相关电子结构的计算和分析。
加深对课堂知识的直观认识,包括能带结构和相关的基本概念等。
二、实验原理第一性原理的理论计算的主要理论基础是量子力学的基本方程和相对论效应,在第一性原理发展过程中,相继提出变分原理、泡利不相容原理、密度泛函理论等。
其基本思路就是它的基本思想,是将多原子构成的实际体系理解为由电子和原子构成的多粒子系统,运用量子力学等基本物理原理最大限度的对问题进行“非经验”处理。
在第一性原理的计算过程中运用了三个近似:非相对论近似(忽略了电子运动的相对论效应);Born-Oppenheimer 近似,核固定近似;单电子近似。
密度泛函理论的主要目标就是用电子密度取代波函数做为研究的基本量。
用电子密度更方便处理。
在密度泛函理论(DFT)中,单电子运动的薛定谔方程按原子单位可表示为)()()()](2[22r k r r V mk k ψεψ=+∇-这里,电荷密度用单电子波函数表示∑=rk r r n 2)()(ψ单电子有效势为)][(′|′r -r |′)r ρ()(ρ93KS ][r V r d r v V xc ++=⎰三、实验内容运用Materials Studio 软件,采用其中的第一性原理计算软件(CASTEP),计算分析不同类型物质(石墨烯、Si、Cu、ZnO)的能带结构、电子态密度和电荷密度。
四、实验步骤1、模型构建建立一个新的project,并在其中建立一个3D工作区域,在菜单栏选择File |Import,显示出Import Document对话框,在对话框中选择Example|Documents |3D model|Si.xsd(硅晶胞模型)并打开,在3D窗口中右击鼠标,选择Display Style,在对话框中选择Ball and stick,并且调节球棍模型尺寸即可得到未修正的硅晶胞原始模型(图1)。
1、castep是用平面波赝势展开波函数,dmol是通过原子轨道的线性组合来处理(castep用的是基于平面波赝势的方法,而dmol是基于分子轨道理论的方法)castep算周期性结构的体系,DMol 适合于分子,团簇,分子筛,分子晶体,聚合物等开放结构。
也就是说对空体积较大的晶体,原子轨道在稀填充体系(原子、分子、团簇、低维周期体系、沸石...)的计算上比平面波有优势。
CASTEP是一个基于密度泛函方法的从头算量子力学程序,总能量包含动能、静电能和交换关联能三部分,各部分能量都可以表示成密度函数。
适用于范围很大的一类固体材料、界面以及表面的性质。
基于总能量的平面波赝势理论,研究的内容包括:结构对称性、晶格参量、键长键角、能带结构、态密度、布局数、光学性能等。
CASTEP 模組允許你使用含有彈性的分子模型工具CASTEP 主要是用於大尺度的週期性系統,他也可以被應用在以超晶格建力起來的缺陷表面介面與分子CASTEP仅能在3D周期模型文件基础上进行计算,必须构建超单胞,以便研究分子体系默认条件下,CASTEP使用得是BFGS几何优化方法,即拟牛顿算法Castep的几何优化过程的本质是期望利用一个迭代过程来完成优化任务,在进行迭代的过程中, 通过调整原子坐标和晶胞参数使结构的总能量最小化。
CASTEP几何优化的核心是通过不断的减小计算力和应力的数量级,直至小于所规定的收敛误差。
当然,也可能给定外部应力张量来对拉应力,压应力和切应力等作用下的体系行为模型化。
在这些情况下反复迭代内部应力张量直到与所施加的外部应力相等Castep默认的能量单位是电子伏特ev,换算关系为1eV=0.036749308Ha=23.0605kcal/mole=96.4853kJ/m2、Energy cutoff 截断能SCF tolerance:迭代标准,就是每两部之间算完的标准( SCF:就是自洽场self consistent field,解薛定谔方程时在开始并不知道波函数,从而也无法获得所需要的电子密度;因此可以先用一个解进行迭代运算,直到最后达到所需要的结果)K point set: K点设置(布里渊区的点数选择,就像你选样本来看产品的合格率一样,选的多就会慢,但会更准确一些)3、建模时加入杂质原子的方法:方法一:用鼠标点上将要被取代得原子(点上后原子颜色将变成黄色),在窗口的右边属性栏中,将会显示这个原子的相关属性,并告诉你这个原子的元素种类(比方是Al 吧),然后点这个元素种类Al,将出现一个元素周期表,选择你要掺杂得原子,确定就可以了!方法二:建立完没有掺杂的晶胞后,选supercell,然后再选择要替换的原子,进行掺杂.如果不把晶胞的对称性改为supercell就不能改变其中的一个原子,而是由于对称性把同一元素的所以原子改变了4、(1)castep建立晶体的模型步骤:build/build crystal,弹出对话框,然后按照晶格参数填入。