无参考基因的转录组分析
- 格式:pdf
- 大小:738.37 KB
- 文档页数:16

2)DNA的质量监测通常有两个方法:首先OD260/OD280比值应该在1.8左右(1.7-1.9),否则意味着DNA样品中存在大量的蛋白质或RNA污染。
其次,琼脂糖电泳分析时应主要以超螺旋条带为主。
最多不超过三条带(分别为超螺旋DNA,线性化DNA和环状DNA)。
否则意味质粒DNA的质量不高,应该重新制备。
2.限制性内切酶的活性1)限制性内切酶一般需要低温保存,而且反复的升降温过程对酶活性的损害很明显。
因而为了确保在有效期内的限制性内切酶不会失活,限制性内切酶的日常保存和使用应当很小。
2)建议购买具有保温功能的冻存盒保存限制性内切酶(-20度),而且取用限制性内切酶时,也应该使用具有保温功能的冻存盒,尽量防止酶的温度反复出现大的波动。
3.限制性内切酶的用量1)限制性内切酶的单位定义通常为:在合适的温度下,完全消化1ugDNA底物所需的酶量定义为一个单位。
2)在这个单位定义中,有几个不确定因素:首先是底物,不同的酶单位定义是选择的底物可能不同(常用的几个底物DNA包括:Lambda DNA ,AD2 DNA 和一些质粒DNA);第二个不确定因素是限制性内切酶在底物DNA上的酶切位点的个数。
由于单位定义中要求完全消化,因而底物上某个酶的酶切位点的个数的多少,就直接影响了该酶的单位定义。
3)因而,在进行酶切时,用1ul酶(一般10IU/ul)消化1ugDNA的通常做法是很不科学的,这也导致在实际工作中,大家要进行多次预实验才能确定最合适酶切条件。
4)以前,我推荐了一个在线的双酶切设计软件,double digestion designer, 可以精确地计算酶切时的限制性内切酶的用量。
使用中,能够注意到,用来进行双酶切的两个酶的用量有时竟然相差近20倍(EcoRI + NheI),而且发现,小片段PCR产物(100-500bp)进行酶切时,需要的酶量比质粒DNA酶切时用量多10倍以上。
5)该软件目前可以免费使用,用户名和密码都是test。

无参考基因的转录组分析无参考基因的转录组分析是指在没有对应基因组序列的情况下,对生物体的转录组数据进行分析,从中获取信息并进行生物学研究。
在无参考基因组的情况下,无法直接对转录组数据进行比对和注释,因此需要采取一些策略和方法来解决这个问题。
1. 转录本组装:通过对转录组数据进行拼接,将转录本组装成单个完整序列,从而获得转录本信息。
这可以使用多个软件来实现,如Trinity、Cufflinks等。
通过对转录本进行定量分析,可以确定各个基因的表达水平。
2. 转录本定量:通过建立转录本的表达矩阵,可以对各个基因的表达水平进行比较和分析。
这可以使用软件如RSEM、eXpress等来完成。
3. 基因功能注释:虽然没有对应基因组序列,但可以利用已知物种的参考基因组信息来进行基因功能注释。
这可以使用一些在线数据库和工具,如Gene Ontology (GO)、KEGG、PANTHER等。
4. 差异表达基因筛选:通过比较不同样本组之间的转录本表达差异,可以筛选出差异表达基因。
这可以使用软件如DESeq2、edgeR等来完成。
5. 寻找新基因:在无参考基因组的情况下,还可以利用转录组数据寻找新基因。
这可以通过比对转录组序列到已知物种的参考基因组上,找出不在参考基因组上的序列,进而预测出新基因。
这可以使用软件如TransDecoder、CPC等来完成。
6.功能富集分析:通过对差异表达基因进行功能富集分析,可以了解这些基因在功能上的特点。
这可以使用一些在线工具和数据库,如DAVID、GSEA等。
7.转录因子分析:转录因子在调控基因的转录过程中起到重要的作用。
通过分析转录因子在转录组中的表达情况,可以了解其在调控过程中的参与情况。
这可以使用一些软件和数据库,如JASPAR、MEME等。
8. 代谢通路分析:通过对差异表达基因进行代谢通路分析,可以了解不同样本组之间在代谢水平上的差异。
这可以使用一些在线工具和数据库,如KEGG、MetaboAnalyst等。

转录组测序以及常用算法简介转录组测序,也被称为“全转录组鸟枪法测序”(WTSS),由于转录组测序的高覆盖率,它也被称为深度测序。
它主要利用新一代高通量测序技术,对物种或组织的RNA反转录而成的cDNA文库进行测序,并得到相关的RNA信息。
其研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
它是指用新一代高通量测序技术,对物种或组织的RNA反转录而成的cDNA文库进行测序,并得到相关的RNA信息。
转录组测序根据有无基因组参考序列分为:有参考基因组的转录组测序,和无参考基因组的de novo测序。
如果有基因组参考序列,可以把转录本映射回基因组,确定转录本位置、剪切情况等更为全面的遗传信息,而这些遗传信息可以广泛应用于生物学研究、医学研究、临床研究中。
虽然转录组测序和基因组测序的步骤大体相同,但是在文库制备和分析方法上却有很大的区别。
在生物信息学领域,序列比对作为识别DNA、RNA和蛋白质相似区域的有效手段,有助于我们更好地研究其结构、功能以及进化方向的关系。
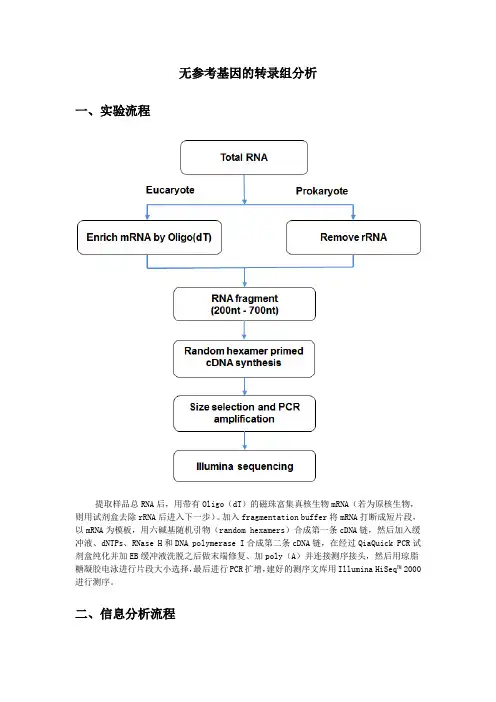
下图简要说明了转录组测序的主要流程:首先将细胞中所有的反转录产物转化为cDNA文库,再将cDNA随机剪切为小DNA片段,并在两端加上接头(Adapter),所得序列通过比对(有参考基因组)或者从头组装de novo(无参考基因组),形成全基因组范围的转录谱。
图1 转录组测序流程图常用算法简介TopHat(/software/tophat/index.shtml)TopHat是Cole Trapnell等人于2009年发表在Bioinformatics上的基于Bowtie的转录组测序比对算法,是马里兰大学生物信息和计算机生物中心,以及加利福尼亚大学伯克利分校数学系和分子细胞生物学系以及哈佛大学的干细胞与再生生物学系联合开发的结果。
它通过超快的高通量短序列比对RNA序列来识别剪切位点。
图2 TopHat流程图TopHat首先先用Bowtie将RNA序列与整个参考基因组进行比对,找到匹配的序列,再用Maq合并匹配的序列,对外显子进行选择性的拼接。

转录组分析学习笔记(持续补充)转录组分析流程(有参和⽆参de novo)1. 获得测序数据,Fastq格式,称之为Raw data。
2. 质量检测3. ⽐对Mapping4. Quantification|Quantitation5. 差异表达分析补充:开始项⽬之前,先确⽴合理的⽂件⽬录结构。
【1】Raw Data 处理理论知识⾼通量测序之所以能够能够达到如此⾼的通量的原因就是他把原来⼏⼗M,⼏百M,甚⾄⼏个G的基因组通过物理或化学的⽅式打算成⼏百bp的短序列,然后同时测序。
在测序过程中,机器会对每次读取的结果赋予⼀个值,⽤于表明它有多⼤把握结果是对的。
从理论上都是前⾯质量好,后⾯质量差。
并且在某些GC⽐例⾼的区域,测序质量会⼤幅度降低。
因此,我们在正式的数据分析之前需要对分析结果进⾏质控。
Fastq ⽂件测序给的“原始数据”,称之为Raw Data。
FASTQ是基于⽂本的,保存⽣物序列(通常是核酸序列)和其测序质量信息的标准格式。
其序列以及质量信息都是使⽤⼀个ASCII字符标⽰,最初由Sanger开发,⽬的是将FASTA序列与质量数据放到⼀起,⽬前已经成为⾼通量测序结果的事实标准。
FASTQ⽂件中以四⾏最为⼀个基本单元,并对应⼀条序列的测序信息,各⾏记录信息如下:第⼀⾏记录序列标识以及相关的描述信息,以‘@’开头,为了保证后续分析软件能够区分每条序列,单个序列的标识必须具有唯⼀性;第⼆⾏为碱基序列;第三⾏以‘+’开头,后⾯是序列标⽰符、描述信息,或者什么也不加;第四⾏,是质量信息,长度和第⼆⾏的序列相对应,每⼀个序列都有⼀个质量评分,根据评分体系的不同,每个字符的含义表⽰的数字也不相同。
碱基质量得分与错误率的换算关系: Q = -10log10p(p表⽰测序的错误率,Q表⽰碱基质量分数)ASCII值与碱基质量得分之间的关系:Phred64 Q=ASCII转换后的数值-64Phred33 Q=ASCII转换后的数值-33⽬前illumina使⽤的碱基质量格式为phred+33, 和Sanger的质量基本⼀致(⽼数据建议查看清楚再进⾏后续处理)。

无参转录组同源基因
无参转录组分析是指在不预先设定任何参考基因或预设条件下,直接对原始转录数据进行全面的分析,以揭示基因表达的内在规律和特征。
同源基因则是指那些在物种间具有相似或相同功能的基因,它们通常在进化过程中保持保守。
无参转录组同源基因分析的目的是识别和比较不同物种或不同组织之间的同源基因,以了解它们在转录水平上的表达模式和功能。
这种分析有助于发现新的生物标记物、药物靶点或疾病相关基因,并深入了解物种间的进化关系和基因功能。
例如,通过无参转录组分析,科学家可以比较不同组织或不同条件下的转录组数据,以发现与特定生理或病理过程相关的同源基因。
进一步的功能研究可以揭示这些基因在相应过程中的作用,并为其潜在的临床应用提供基础。
总之,无参转录组同源基因分析是一种强大的方法,用于深入了解基因表达的内在规律和特征,以及物种间的进化关系和基因功能。
这种分析有助于发现新的生物标记物、药物靶点或疾病相关基因,并为未来的生物医学研究提供有价值的线索。

无参转录组分析结果的解读近年来,转录组技术被广泛用于研究基因组功能,而无参转录组(RNA-seq)是用于转录组分析的一种测序技术,其核心原理是通过链特异性加速器(CLASP)进行高通量测序,从而实现对大量基因表达的实时检测,获取转录组的精准分析结果。
无参转录组分析是一个复杂的系统,它结合了基因表达、结构和功能的方面,以及基因之间的分子关系,以便深入的解释和评估基因的表达和调控机制。
无参转录组分析可以协助研究者分析品系和表达模式之间的差异,用于研究基因之间的联系与基因网络,指导药物研发等多方面。
无参转录组分析方法由若干部分组成,主要分为基因表达、基因功能分类和网络功能分析三个步骤。
第一步是基因表达,通过测序产生的数据,采用RPM和FPKM等指标,计算基因表达水平,以定量检测基因在不同样本中的表达变化。
其次是基因功能分类,通过特定算法,检索基因具有特定功能的蛋白质序列,进而确定基因所处细胞环境,了解不同表达基因在特定环境中发挥的作用。
最后是网络功能分析,使用聚类分析,结合元数据构建基因相互作用和调控网络的全貌,以及网络中的细胞因子和调控子的联系,从而可以更加深入的了解基因的表达与功能之间的关系。
无参转录组分析可以提供全面而准确的表达信息,以及基因之间的联系,可以应用于多种领域,如病原学研究、疾病免疫学检测、细胞功能的解析、精准医学建模和药物研发等。
但是,所获得的结果是相对的,必须根据不同的试验要求、受检对象和基因组的不同,以及参考的数据的质量等,综合考虑后才能做出准确的解释和评估。
总之,无参转录组分析技术能够获得准确有效的基因表达分析结果,为生物学研究以及药物研发等提供有力的支持,但也需要对技术、参考数据和评估细节都要进行严谨的评估,才能得到准确的结果。
此外,无参转录组分析的可行性取决于参与者的资源和技术水平,技术突破以及良好的生物组学等应用水平,可以有效提升无参转录组分析的准确性和可靠性,有利于更好的解读分析结果。

利用转录组测序分析大豆矮小突变体中差异表达基因近年来,随着测序技术的发展,转录组测序成为研究生物体内基因表达的重要手段之一。
通过转录组测序,可以得知特定生物体在某个生长阶段或环境适应中的基因表达情况。
在大豆研究中,通过转录组测序研究矮小突变体的差异表达基因,可以帮助我们深入了解大豆生长发育中的分子机制。
矮小突变体是指生长期较同个物种的正常个体矮小的一类突变体。
在大豆中,矮小突变体的发现对于提高大豆的产量和抗病能力具有重要意义。
通过转录组测序研究矮小突变体中的差异表达基因,可以揭示矮小突变体生长发育的分子机制,从而为大豆育种提供理论依据。
在利用转录组测序研究大豆矮小突变体中的差异表达基因时,首先需要选择合适的实验材料和对照材料。
一般情况下,可以选择与突变体具有相似生长发育阶段的野生型作为对照材料。
然后,通过高通量测序技术对突变体和对照样品进行测序,获取大量的转录组测序数据。
接下来,对测序数据进行质量控制和过滤,去除低质量的数据和接头序列。
然后使用比对算法将测序数据比对到参考基因组上,得到每个基因的表达情况。
通过比较突变体和对照样品之间的差异表达基因进行筛选,并对其进行功能注释和富集分析。
差异表达基因分析的结果通常可以揭示突变体和对照样品在基因表达水平上的差异,可以给出候选基因的列表。
我们可以进一步对这些候选基因进行生物学实验验证,以了解这些基因在大豆生长发育中的具体功能。
通过转录组测序研究矮小突变体中的差异表达基因,可以明确突变体生长发育过程中哪些基因发生了变化,从而了解突变体的生长发育机制。
此外,通过功能注释和富集分析,我们还可以了解突变体中差异表达基因所参与的生物学过程和通路。
转录组测序研究在大豆育种中的应用前景广阔。
通过研究矮小突变体中差异表达基因,可以为大豆产量和抗病能力的提高提供重要的理论基础。
同时,转录组测序还可以帮助我们发现更多的潜在基因,用于大豆的遗传改良和功能研究。
总之,利用转录组测序研究大豆矮小突变体中的差异表达基因有助于我们了解大豆生长发育过程中的分子机制。

送样要求动植物基因组从头测序1、DNA样品:基于Illumina平台,PE文库DNA浓度≥20 ng/μl,总量≥6μg(荧光定量),MP文库DNA 浓度≥40ng/μl,总量≥12μg(荧光定量);基于Roche 454 FLX+ 平台,DNA浓度≥20ng/μl,总量≥3μg(荧光定量),电泳检测无明显RNA条带,基因组条带清晰、完整,主带应在100 kb以上。
若样品中有多糖、糖蛋白的残留,对打断DNA样品带来非常大的困难,且很难去除,因此特别要求所提供的样品不要有多糖或糖蛋白污染。
2、动物样品:样品最好来自纯系,对于一般物种应挑选肝脏、肾脏、血液等组织取样,对于珍贵物种请提供耳样、毛发(带毛根)等脂肪含量较少的组织进行取样。
为了减少个体差异对后续拼接产生的影响,尽量从同一个个体中取样。
若物种体积较小,从一个个体中提取的DNA量不能满足测序实验所需,在保证量的前提下,应尽量减少采样个体的数量。
提供组织样品应>500mg,尽量提供较多量,不用的物种DNA 提取产物有差异。
3、植物样品:样品最好来自纯合体或单倍体。
需为黑暗无菌条件下培养的黄化苗或组织样品。
提供组织样品应>500mg,尽量提供较多量,不同的物种DNA提取产量有差异动植物基因组重测序1、DNA样品:Miseq DNA PE 文库浓度≥20 ng/μl,总量≥6μg(荧光定量);Miseq DNA MP文库浓度≥40ng/μl,总量≥12μg(荧光定量);454 DNA库浓度≥20ng/μl,总量≥3μg(荧光定量),电泳检测无明显RNA条带,基因组条带清晰、完整,主带应在100 kb以上。
若样品中有多糖、糖蛋白的残留,对打断DNA样品带来非常大的困难,且很难去除,因此特别要求所提供的样品不要有多糖或糖蛋白污染。
2、动物样品:对于一般物种应挑选肝脏、肾脏、血液等组织取样,对于珍贵物种请提供耳样、毛发(带毛根)等脂肪含量较少的组织进行取样。

生物信息学中转录组学数据分析的方法与工具转录组学是研究基因组中所有转录本的总体表达情况的一项重要分析技术。
随着高通量测序技术的发展,转录组学数据的分析在解析物种的转录调控、功能基因和代谢途径等方面发挥了关键作用。
本文将介绍生物信息学中转录组学数据分析的常用方法与工具。
首先,对于转录组学数据的分析,首先要进行质量控制。
质量控制可以帮助鉴定测序过程中的技术偏差和样本质量问题。
常用的质量控制工具包括FastQC和Trim Galore。
FastQC可以通过对测序数据进行质量评估,提供关于测序质量、GC 含量、碱基分布和测序片段长度等信息。
Trim Galore则可以根据FastQC结果进行质量修剪,去除低质量碱基和接头序列。
接下来的步骤是进行序列比对。
比对是将测序reads映射到参考基因组上的过程。
在转录组学数据中,常用的比对工具包括Bowtie、TopHat、STAR和HISAT 等。
Bowtie是一款快速比对工具,可用于对短序列的比对。
TopHat则是用于剪接位点的比对,可以识别剪接事件。
STAR和HISAT则是新一代快速比对工具,可以同时比对测序reads和剪接位点。
完成比对后,接下来要对比对结果进行定量。
转录组学数据的定量常用的方法有基于基因表达量和基于转录本表达量两种方式。
基于基因表达量的分析可以直接对比对到基因组的reads数量进行统计,常用的工具包括HTSeq和featureCounts。
基于转录本表达量的分析则可以将reads根据转录本注释进行分配,常用的工具包括Cufflinks和StringTie等。
在定量完成后,我们可以进行差异表达基因分析。
差异表达分析可帮助我们找到在不同组之间表达水平差异显著的基因。
常用的差异表达基因分析工具有DESeq2、edgeR和limma-voom等。
这些工具可以从统计学的角度评估差异表达的可靠性,并提供一系列的统计分析方法和可视化工具。
此外,转录组数据的富集分析也是转录组学数据分析的重要部分。
高通量转录组测序的数据分析与基因发掘周华;张新;刘腾云;余发新【摘要】高通量转录组测序(RNA-seq)是在转录组水平上进行深度测序的一项技术,为真核生物转录组学的研究开创了新平台,但同时测序所得到的海量数据的生物信息学分析成为科研工作者的一大挑战。
对转录组测序技术进行了阐述,重点介绍了转录组测序后的数据分析,以及在真核生物尤其是非模式物种中的基因发掘方法。
%High-throughput transcriptome sequencing (RNA-seq) is a recently developed approach to transcriptorne profiling that uses deep-sequencing technologies. It provided a novel platform for eu- karyotie transeripome researches, but the bioinformatics analysis of sequencing data became the chal- lenge of scientific worker. In this review, we described the researches process of high-throughput transcriptome sequencing technology, focusing on sequencing data analysis and gene discovery of dif- ferent species, especially of non-model species.【期刊名称】《江西科学》【年(卷),期】2012(030)005【总页数】5页(P607-611)【关键词】转录组测序;数据分析;基因发掘【作者】周华;张新;刘腾云;余发新【作者单位】江西省科学院生物资源研究所,江西省观赏植物遗传改良重点实验室,江西南昌330029;南京市林业站,江苏南京210036;江西省科学院生物资源研究所,江西省观赏植物遗传改良重点实验室,江西南昌330029;江西省科学院生物资源研究所,江西省观赏植物遗传改良重点实验室,江西南昌330029【正文语种】中文【中图分类】Q987转录组研究是一个发掘功能基因的重要途径,是基因功能及结构研究的基础和出发点。
RNA-seq 名词解释诺禾致源转录调控研究部2014.03.21基本概念RNA-seq:基于二代测序技术,研究特定细胞在某一功能状态下所有RNA的功能,主要包括mRNA和非编码RNA。
能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息,已广泛应用于基础研究、临床诊断和药物研发等领域。
Q20,Q30:Phred 数值大于20、30的碱基占总体碱基的百分比,其中Phred=-10log10(e).gene:具有编码蛋白质或决定某一性状作用的一段核酸序列。
intron:内含子,是真核生物细胞DNA中的间插序列。
这些序列被转录在前体RNA中,经过剪接被去除,最终不存在于成熟RNA分子中。
术语内含子也指编码相应RNA内含子的DNA中的区域。
exon:外显子,是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。
外显子是最后出现在成熟RNA中的基因序列,又称表达序列。
既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。
术语外显子也指编码相应RNA外显子的DNA中的区域。
intergenic:基因间区,指基因与基因之间的间隔序列,不属于基因结构,不直接决定氨基酸,可能通过转录后调控影响性状的区域。
UTR:Untranslated Regions, 非翻译区域。
是信使RNA(mRNA)分子两端的非编码片段。
5'-UTR从mRNA起点的甲基化鸟嘌呤核苷酸帽延伸至AUG起始密码子,3'-UTR从编码区末端的终止密码子延伸至多聚A尾巴(Poly-A)的前端。
transcript:转录本,是由一条基因通过转录形成的一种或多种可供编码蛋白质的成熟的mRNA。
一条基因通过内含子的不同剪接可构成不同的转录本。
isoform:同一个基因经可变剪切或内含子选择机制产生不同的转录本,这些不同转录本即称isoform。
无参转录组之转录本质控今天是生信星球陪你的第267天大神一句话,菜鸟跑半年。
我不是大神,但我可以缩短你走弯路的半年~就像歌儿唱的那样,如果你不知道该往哪儿走,就留在这学点生信好不好~这里有豆豆和花花的学习历程,从新手到进阶,生信路上有你有我!豆豆写于19.2.2之前写了无参转录组知识储备、无参转录组起步今天主要看看转录本拼接质量评估前言我们利用trinity简单的命令拼接好转录本后,想要知道我们拼接的质量如何,如果不好可能需要寻找原因,重新调整参数再重新拼接。
这一步是很关键的,因为拼接的转录本相当于有参里面的参考转录组,试想如果参考都做不好,那岂不是上梁不正下梁歪?首先我们可以看看组装了多少条转录本grep '>' $wkd/assembly/trinity_out_dir/Trinity.fasta | wc -l但这仅仅是最粗略的方式,因为随着测序深度的加大,得到的contigs越多,因此可以拼接更多的转录本另外可以看看基本的统计值TrinityStats.pl $wkd/assembly/trinity_out_dir/Trinity.fasta第一部分Counts of transcripts, etc. 结果中的Total trinity transcripts 和上面那个命令结果一致【正常数据中转录本数量小于20w是正常的,如果数量达到了30w、40w条,就需要先用corset软件进行聚类】结果中还有组装的gene数Total trinity 'genes' ,可以看到transcripts的数量比genes的数量多,因为存在一个基因的可变剪切(这种情况在昆虫和哺乳动物中比较常见)另外还有第二部分内容Stats based on ALL transcript contigs: N50值(at least half of the assembled bases are in contigs of at least that contig length累加后长度超过转录组总长度一半的contig的长度就是N50)【正常情况下,N50应该是1k左右】说到评估,我们一般会想到:和其他拼接工具的结果进行对比(就好像在有参中使用3-5中工具进行比对,看看分别的比对率),或者使用不同的参数看看结果异同(前提是对参数的设置比较了解)总的来说,上面的方法得到的结果不是特别有用,有下面几种方法可以更有效地评价转录本质量第一种 align将reads重新比对到转录本上,看看比对率一般来讲,至少应该有80%的原始数据可以在拼接的转录本中找到(经验值),剩下的没有拼接上的序列可能由于低表达导致没有足够的覆盖度进行拼接或者序列质量较低或者重复reads。
染色质免疫共沉淀技术(Chromatin Immunoprecipitation,ChIP)也称结合位点分析法,是研究体内蛋白质与DNA相互作用的有力工具,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。
将ChIP与第二代测序技术相结合的ChIP-Seq技术,能够高效地在全基因组范围内检测与组蛋白、转录因子等互作的DNA区段。
ChIP-Seq的原理是:首先通过染色质免疫共沉淀技术(ChIP)特异性地富集目的蛋白结合的DNA片段,并对其进行纯化与文库构建;然后对富集得到的DNA片段进行高通量测序。
研究人员通过将获得的数百万条序列标签精确定位到基因组上,从而获得全基因组范围内与组蛋白、转录因子等互作的DNA区段信息。
技术路线实验流程生物信息分析流程测序对客户提供的ChIP样品(如果有阴阳参启动子区域或DNA序列的)进行定量检测,检测合格后进行测序文库构建、DNA成簇(Cluster generation)扩增、高通量测序。
基本数据分析数据产出统计:对测序结果进行图像识别(Base calling),去除污染及接头序列;统计结果包括:测定的序列(Reads)长度、Reads数量、数据产量。
高级数据分析标准高级数据分析内容包括:(1)ChIP-Seq序列与参考序列比对;(2)Peak calling:统计样品Peak信息(峰检测及计数、平均峰长度、峰长中位数);(3)统计样品Uniquely mapped reads在基因上、基因间区的分布情况及覆盖深度;(4)给出每个样品Peak关联基因列表及GO功能注释;(5)在多个样品间,对与Peak关联基因做差异分析。
转录组基因分析:RNA-seqRNA-seq即转录组测序技术,就是把mRNA,smallRNA,andNONcoding RNA等或者其中一些用高通量测序技术把它们的序列测出来。
反映出它们的表达水平。
转录组是某个物种或者特定细胞类型产生的所有转录本的集合。
我这种情况,做转录组应该选无参还是有参?先说下结论吧,建议按无参转录组去做,因为50%的回比率实在是太低了,一般来说起码也得达到60%-65%。
按有参去做会浪费很多的数据,而且会有很多基因不会被检测到。
下面是我们之前写的一个关于转录组分析模式的选择建议,希望对你有所帮助。
同样是用NGS方法做转录组,根据测序物种和实验目的的不同,在分析模式上会有差别,因此了解不同分析模式及其适用范围对于选择适合自己实验的分析方法有重要的意义。
三种转录组分析模式基于研究物种是否有参考基因组,实验目的上是否需要分析新的转录本,转录组测序的分析模式大致可以分成3种类型,如下图:一、有参,需要分析新转录本部分有参考基因组的物种,由于注释信息不够完善,或需要分析一些非编码RNA,这时需要基于Reads与基因组比对信息对转录组进行组装,以期获得新的转录本来让分析结果更加完备。
这也是有参物种做转录组最常用的分析模式,其分析步骤如下:1. Reads与基因组比对2. 基于比对信息组装转录本3. 基因或转录本表达定量4. 差异分析和功能富集分析二、有参,只分析已知转录本针对参考基因组注释信息较为详细的物种,比如人,小鼠,拟南芥等模式生物,同时您的实验目的很明确,就是分析已知的基因或转录本,那就可以直接基于基因组注释信息中提取出的转录本序列来进行后续分析。
该分析模式分析流程简单、速度快,其具体分析步骤如下:1. Reads与转录本序列进行比对2. 转录本表达定量3. 差异分析和功能富集分析三、无参考基因组的转录组而对于没有参考基因组的物种,或者基因组组装不好的物种,必须先使用测序数据组装一套转录本,再基于转录本进行后续分析。
其分析步骤如下:1. Reads De novo组装转录本序列2. Reads 回比组装好的转录本序列3. 转录本表达定量4. 差异表达分析和功能分析。