PDB文件详解
- 格式:doc
- 大小:28.50 KB
- 文档页数:2

pdb跳出函数PDB是一种调试信息文件,它包含了程序的符号表、源代码行号、变量名等信息,可以帮助程序员在调试时更快地定位问题。
在调试过程中,我们经常需要跳出函数来查看当前的变量值、调用栈等信息,这时候就需要用到PDB跳出函数。
PDB跳出函数的实现方法有很多种,下面介绍一种比较常用的方法:首先,我们需要在代码中插入一个断点,可以使用Visual Studio等IDE自带的断点功能,也可以使用DebugBreak()函数来手动插入断点。
当程序执行到断点处时,会暂停执行,等待我们进行调试操作。
接下来,我们需要打开PDB文件,可以使用Visual Studio等IDE自带的调试工具,也可以使用WinDbg等第三方调试工具。
在PDB文件中,我们可以查看当前函数的符号表、源代码行号等信息,也可以查看当前线程的调用栈、变量值等信息。
然后,我们需要跳出当前函数,可以使用Step Out等调试命令来跳出函数。
跳出函数后,我们可以查看当前函数的返回值、调用栈等信息。
最后,我们可以继续执行程序,直到下一个断点处或程序结束。
在程序执行过程中,我们可以随时使用PDB跳出函数来查看当前的调试信息,帮助我们更快地定位问题。
总的来说,PDB跳出函数是调试过程中非常重要的一个工具,它可以帮助我们更快地定位问题,提高调试效率。
在使用PDB跳出函数时,我们需要注意以下几点:1. 在插入断点时,要选择合适的位置,避免影响程序的正常执行。
2. 在查看PDB文件时,要注意当前线程的上下文环境,避免查看到错误的信息。
3. 在跳出函数时,要注意当前函数的返回值和调用栈,避免出现错误的结果。
4. 在程序执行过程中,要随时注意程序的状态,避免出现意外情况。
综上所述,PDB跳出函数是调试过程中非常重要的一个工具,它可以帮助我们更快地定位问题,提高调试效率。
在使用PDB跳出函数时,我们需要注意以上几点,避免出现错误的结果。

pdb 用法
PDB(Protein Data Bank)是一个存储生物大分子(如蛋白质、核酸等)结构信息的数据库。
PDB 文件包含有关分子的三维坐标、结构信息、生物学相关的元数据等。
以下是一些常见的 PDB 文件的用法:
查看 PDB 文件:
PDB 文件通常是文本文件,你可以使用文本编辑器查看其内容。
例如,使用命令行下的 cat(Linux/macOS)或 type(Windows):cat your_file.pdb
分析 PDB 文件:
你可以使用专业的生物信息学工具来分析PDB 文件,例如BioPython、PyMOL、或者 Rosetta。
这些工具提供了丰富的功能,用于解析、可视化、分析生物大分子的结构。
PDB 文件格式:
如果你想编写脚本来处理PDB 文件,了解PDB 文件的格式是很重要的。
PDB 文件的格式描述了原子的坐标、结构信息等。
详细了解 PDB 文件格式可以帮助你更好地处理和解析数据。
获取 PDB 文件:
你可以从PDB 数据库或其他相关数据库中下载PDB 文件。
在网站上搜索感兴趣的分子或结构,并下载相应的 PDB 文件。
可视化 PDB 文件:
使用专业的分子可视化工具如 PyMOL、VMD 或者 Chimera,可以加载 PDB 文件并以三维方式展示生物大分子的结构。
请根据你的具体需求选择适当的工具和方法。
生物信息学领域有很多工具和资源,适用于不同的任务和分析。
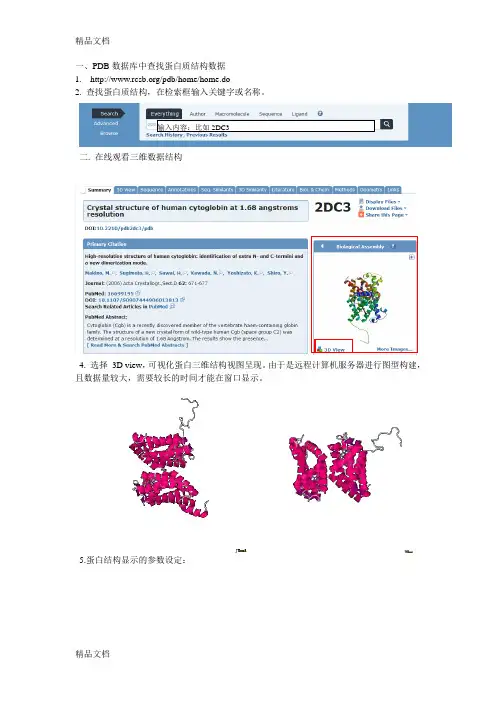
一、PDB数据库中查找蛋白质结构数据
1./pdb/home/home.do
2. 查找蛋白质结构,在检索框输入关键字或名称。
输入内容:比如2DC3
二. 在线观看三维数据结构
4. 选择3D view,可视化蛋白三维结构视图呈现。
由于是远程计算机服务器进行图型构建,且数据量较大,需要较长的时间才能在窗口显示。
自定义设定
脚本选项:
二、下载三维数据文件
1. 在查询结果窗口的右边找到以下图型。
如图:选择Download Files。
2.确定下载的文件类型,一般根据你要使用蛋白质结构数据而定。
选择PDB File(Text).
3. 用写字板打开PDB文件,可以看到蛋白质数据库文件实质也是文本文件。
4. 下载Pymol,并安装。
点DOWNLOAD进入下载,选择windows版本,并安装(需要pay money)。
或到此网站下载:/~gohlke/pythonlibs/#pymol
选择win32,1.7.1.1版本。
并安装,需要python编程软件的支持,可从360软件管件直接安装。
安装如图:
使用详情参见教程,注意打开程序需要找到python的安装目录:C:\Python27\PyMOL,找到PyMOL.exe文件,双击启动程序。
打开2DC3.pdb
或使用RasMol软件进行三维视图分析,步骤同上。

PDB⽂件详解1. PDB⽂件的介绍PDB(Program Data Base),意即程序的基本数据,是VS编译链接时⽣成的⽂件。
DPB⽂件主要存储了VS调试程序时所需要的基本信息,主要包括源⽂件名、变量名、函数名、FPO(帧指针)、对应的⾏号等等。
因为存储的是调试信息,所以⼀般情况下PDB⽂件是在Debug 模式下才会⽣成。
2. PDB⽂件的调⽤过程模块(Module),EXE和DLL都可以称之为模块,因为它们都有⾃已独⽴的Stack,所以我们在调试程序时,可以在Call Stack窗⼝查看到所有调⽤的Module Name。
并且可以右键查看相应模块的ybmol Load Information,即该模块调⽤的PDB⽂件路径的过程。
每个模块被载⼊的时候,其相同名字的PDB⽂件同时被载⼊。
所以Debug模式下,不仅因为代码没有优化,同时因为要载⼊PDB⽂件,所以Debug模式下的程序执⾏速度⾮常慢。
每个模块只会⽣成⼀个相同名字的PDB⽂件,并且模块⽣成的同时,会校验PDB⽂件⽣成GUID记录在模块内。
这是因为调试时,调试器强制要求每个模块必须和PDB⽂件保持⼀致。
实验过程中,⽤之前⽣成的PDB⽂件替换当前⽣成的PDB⽂件时,Debug窗⼝会显⽰No symbols loaded. MSDN也做了相应的说明:The debugger will load only a PDB for a binary that exactly matches the PDB that was created when the binary was built.PDB⽂件中记录了源⽂件路径的相关信息,所以在载⼊PDB⽂件的时候,就可以将相关调试信息与源码对应。
这样可以可视化的实时查看调试时的函数调⽤、变量值等相关信息。
模块当中记录的PDB⽂件是绝对路径。
所以只要模块在当前电脑上载⼊,调试器⾃然地会根据模块当中的路径信息找到相应PDB⽂件并载⼊。

水分子的pdb格式水分子(H2O)是地球上最常见的分子之一,也是生命存在所必需的物质之一。
它的pdb格式是一种常用的文件类型,能够描述水分子的空间结构和化学性质。
本文将为大家介绍水分子的pdb格式,并探讨其重要性以及在生物科学研究中的应用。
首先,让我们来了解一下pdb格式。
PDB是Protein Data Bank的缩写,是一种用于存储蛋白质和其他生物大分子结构数据的文件格式。
水分子的pdb格式通常包含了水分子中每个原子的坐标和相关信息,以及水分子与其他分子之间的相互作用。
通过这种格式,科学家们可以准确地描述水分子的组成和结构,以及水分子在溶剂中的行为。
水分子的pdb格式具有生动的特点,可以帮助我们更好地理解水分子的结构和性质。
在pdb文件中,水分子通常由两个氢原子和一个氧原子组成,呈现出一个V字形的结构。
氧原子位于两个氢原子之间,成为水分子的核心。
氧原子带有部分负电荷,而氢原子则带有部分正电荷,这使得水分子呈现出极性。
水分子的极性使其具有许多重要的性质和应用。
首先,由于水分子的偏极性,它可以与其他极性分子或离子发生氢键作用,这种作用对于生物大分子的稳定性和功能至关重要。
此外,水分子还具有优良的溶剂能力,能够溶解许多物质,包括离子、极性分子和非极性分子。
这使得水成为生物体内许多化学反应和生物过程的重要媒介。
在生物科学研究中,pdb格式的水分子数据广泛应用于蛋白质结构研究、分子模拟和药物设计等领域。
通过分析水分子在蛋白质中的位置和相互作用,科学家们可以揭示蛋白质的结构和功能机制。
此外,水分子还被用来模拟和预测生物大分子的稳定性和互动行为,为药物设计和开发提供重要的理论基础。
总结起来,水分子的pdb格式是一种描述水分子结构和性质的文件格式,具有生动、全面和指导意义的特点。
它不仅帮助我们理解水分子的组成和结构,还对生物科学研究起到重要的作用。
通过深入研究水分子的pdb格式,我们可以更好地认识水的重要性和多样性,进一步推动科学的发展和应用。

pdb数据库使用方法PDB数据库(Protein Data Bank)是一个著名的生物科学数据库,收录了各种生物大分子三维结构的信息。
本文将介绍PDB数据库的使用方法,帮助读者更好地利用这个有用的资源。
一、了解PDB数据库及其结构PDB数据库是由许多研究机构、大学和政府机构建立和维护的,收录了全球范围内各种生物大分子的三维结构数据,如蛋白质、核酸和复合物等。
PDB数据库中每一项数据都对应一个唯一的PDB ID号码,并且提供了该生物大分子的结构信息、序列信息、实验条件及解析方法等详细的数据。
为方便使用,PDB数据库的数据以PDB格式存储。
二、使用PDB数据库的搜索功能PDB数据库提供了一系列搜索选项,让用户按需查询数据。
用户可以按照PDB ID、蛋白质名字、结晶学条件等多种方式进行搜索。
在PDB数据库的主页页面,用户可以看到搜索选项,点击“Search”按钮即可进入搜索页面进一步操作。
三、使用PDB格式数据文件PDB格式数据文件是PDB数据库中唯一的数据类型,存储了生物大分子的结构信息、序列信息等所有数据。
这些数据可以通过下载PDB文件的方式进行获取。
用户可以在PDB数据库中找到对应的数据,进入数据详情页面后,点击“Download Files”按钮即可下载PDB格式的文件。
四、使用PDB格式数据文件的软件PDB格式的数据文件可以在很多软件上进行解析和编辑,包括众所周知的PyMol、Chimera等生物大分子分析软件,也有很多其他免费的软件可以用来查看或编辑PDB文件,如UCSF ChimeraX、MSM格式转换器等。
用户可以根据自己的需要选择合适的软件进行使用。
五、常用生物大分子分析软件介绍1. PyMolPyMol是一款非常流行的分子可视化软件,用于可视化、分析和编辑生物大分子的三维结构。
该软件具有强大的分子动画和交互式残基操作功能,可以进行序列对齐、氨基酸置换等功能,适合于研究生物分子结构和功能。

atp小分子pdb格式
ATP(三磷酸腺苷)是一种重要的生物分子,其PDB格式是一种
常见的用于描述蛋白质和其他生物大分子结构的文件格式。
PDB格
式是一种文本文件格式,用于存储生物大分子的结构信息,包括原
子坐标、拓扑信息和晶体学数据等。
在PDB格式中,每个原子的坐
标和化学信息都有其特定的位置和格式。
对于ATP的小分子结构,其PDB格式文件将包含有关ATP分子
的原子坐标、键的连接信息、结合的水分子以及可能的离子等信息。
PDB文件中还可能包含与ATP分子相关的晶体学数据,如晶胞参数、空间群信息等。
从PDB格式文件中可以获取ATP分子的三维结构信息,这对于
研究ATP与其他生物大分子(如蛋白质)的相互作用、药物设计等
具有重要意义。
研究人员可以使用PDB格式文件中的信息进行分子
建模、动力学模拟、结构比对等操作,从而深入了解ATP分子的结
构与功能。
总之,ATP小分子的PDB格式文件是一种重要的数据资源,可
以为生物化学和药物研究提供宝贵的结构信息。
通过对PDB文件中
的信息进行分析和处理,研究人员可以更好地理解ATP分子的结构特征及其在生物体内的生物学功能。

蛋白三级结构pdb文件简介蛋白是生物体内的重要组成部分,它们扮演着许多关键生物功能的角色,比如催化化学反应、传递信号和维持细胞结构等。
了解蛋白的结构对于理解其功能以及设计新药物等具有重要意义。
在蛋白领域,pdb文件被广泛用于表示蛋白的结构信息。
本文将深入探讨蛋白三级结构pdb文件的相关知识。
三级结构和pdb文件的定义三级结构蛋白的三级结构是指蛋白质链上不同部分的折叠方式和排列顺序。
它由多个二级结构(α-螺旋,β-折叠等)组成,同时还包括各个二级结构之间的连接和排列方式。
三级结构对于蛋白的功能和稳定性至关重要。
pdb文件pdb文件是蛋白三级结构的标准文件格式,它以文本形式存储蛋白的结构信息。
每个pdb文件对应一个蛋白质,其中包含了描述其结构的坐标和拓扑信息。
pdb文件的内容包括蛋白的原子坐标、残基标识、结构域等信息。
pdb文件的组成HEADERHEADER记录包含了pdb文件的描述信息,比如标题、蛋白质名称等。
ATOMATOM记录包含了蛋白质中原子的坐标信息。
每一行都代表了一个原子的位置,包括其X、Y、Z坐标以及元素类型等。
HETATMHETATM记录包含pdb文件中非蛋白原子的信息,比如水分子、小分子配体等。
SEQRESSEQRES记录包含了蛋白质的氨基酸序列信息。
它标识出了每个结构域中的残基编号和对应的氨基酸类型。
CONNECTCONNECT记录包含了蛋白质原子之间的连接信息。
它描述了非共价键的连接关系,比如氢键、离子键等。
MASTERMASTER记录包含了pdb文件的整体信息,比如原子总数、链的数量等。
ENDEND标志着pdb文件的结束。
pdb文件的解析和应用解析pdb文件的工具解析pdb文件需要使用特定的工具和库,比如Bio.PDB库。
该库提供了用于解析pdb文件的各种函数和方法,可以轻松地读取pdb文件并提取相关的结构信息。
pdb文件的应用pdb文件在蛋白领域有广泛的应用。
它们可以用于蛋白结构预测、药物设计和生物大数据分析等方面。

pdb⽂件⼩结.pdb⽂件,是VS⽣成的⽤于调试的符号⽂件(program database),保存着调试的信息。
在VS的⼯程属性,C/C++,调试信息格式,设置/Zi,那么VS就会在构建项⽬时创建PDB⽂件。
在这⾥要区分两种情况:1、构建静态库时,可以在⼯程属性 –> C/C++ –> 输出⽂件 –> 程序数据库名设置⽣成的pdb⽂件名称,如果不指定,默认是⽣成为VCx0.pdb,这⾥x是VS版本号,例如⽤VS2005,就会⽣成VC80.pdb。
这⾥就会产⽣⼀个疑问,编译静态库时默认⽣成的.pdb⽂件名字都⼀样,那引⽤这个静态库的项⽬最后能找到正确的.pdb⽂件吗?答案是肯定的,因为VS会在⽣成的⽂件中嵌⼊ .pdb ⽂件的路径。
举个例⼦,在Project/ToolA下,构建了⼀个静态库ToolA.lib,对应⽣成⼀个vc80.pdb,同样在在Project/ToolB下,构建了⼀个静态库ToolB.lib,对应⽣成⼀个vc80.pdb。
然后最终的⼯程Work.exe同时链接了这两个静态库.这时,⽣成Work.pdb的时候,就会在ToolA.lib中找到它对应的符号⽂件路径Project/ToolA/vc80.pdb,以及ToolB.lib对应的符号⽂件路径Project/ToolB/vc80.pdb,合并⽣成最终⼯程的Work.pdb。
2、构建可执⾏⽂件或动态库,这种情况下,编译器会⽣成⼀个.pdb⽂件,链接器会⽣成⼀个.pdb⽂件,编译器⽣成的pdb⽂件可以在在⼯程属性 –> C/C++ –> 输出⽂件 –> 程序数据库名设置,链接器⽣成的.pdb⽂件可以在⼯程属性 –> 链接器 –> 调试 –> ⽣成调试信息(设置Yes),⽣成程序数据库名设置。
这两个pdb⽂件有什么不⼀样呢?编译器⽣成的pdb⽂件,默认也是⽤vcx0命名,是编译器在编译过程中,把每个.obj⽂件对应的符号信息存储在其中的,但不包括函数定义。

PDB文件详解PDB格式文件对大部分做模拟和计算的人来说都很熟悉,但其中各个参数的意义很多人并不是很了解。
从网上搜集了一些文章,结合自己的知识来对PDB文件中各个参数的意义做个解释:REMARK该记录用来记述结构优化的方法和相关统计数据。
如用Refmac进行结构优化,该记录将自动插入输出的PDB。
CRYST1 (NMR除外)该记录用来记述晶胞结构参数(a, b, c, α, β, γ, 空间群) 以及Z值(单位结构中的聚和链数)。
SCALEn(n = 1, 2, 3) (NMR除外)该记录介绍数据中直角坐标向部分晶体学坐标的转换。
ATOM该记录记述了标准氨基酸以及核酸的原子名,残基名,直角坐标,占有率,温度因子等信息。
HETATM该记录记述了标准氨基酸以及核酸以外的化合物的原子名,残基名,直角坐标,占有率,温度因子等信息。
TER该记录表示链的末端。
在每个聚合链的末端都必须有TER记录,但是由于无序序列而造成的链的中断处不需要该记录。
MODEL当一个PDB文件中包含多个结构时(例:NMR结构解析),该记录出现在各个模型的第一行。
MODEL记录行的第11-14列上记入模型序号。
序号从1开始顺序记入,在11-14列中从右起写。
比如说有30个模型,则第1至9号模型,该行的7-13列空白,在14列上记入1-9的数字;第10-30号模型,该行的7-12列空白,13-14列上记入10-30的数字。
ENDMDL与MODEL记录成对出现,记述在各模型的链末端的TER记录之后。
END该记录标志PDB文件的结束,是必需的记录。
B-factoerThe B-factor (or temperature factor) is an indicator of thermal motion about an atom. However, it should be pointed out that the B-factor is a mix of real thermal displacement, static disorder (multiple but defined conformations) and dynamic disorder (no defined conformation), and all the overlap between these definitions.是晶体学中的一个重要参数,晶体学中结构因子可以表达为坐标x , y, z与Bj 因子的函数。
蛋白三级结构pdb文件蛋白三级结构是指蛋白质分子的空间构象,包括原子的位置和它们之间的相互作用。
PDB文件是蛋白质三级结构的标准格式,它包含了蛋白质的原子坐标、拓扑信息以及其他相关信息。
下面将介绍PDB文件的结构和内容。
1. PDB文件格式PDB文件是一种文本文件,通常以.pdb为扩展名。
它由多行组成,每行最多包含80个字符。
PDB文件中的每个原子都有一个唯一的标识符,称为原子序号(ATOM序号)。
ATOM序号由6个字符组成,前4个字符是ATOM,后面2个字符是序号,从1开始递增。
2. PDB文件内容PDB文件包含了蛋白质的原子坐标、拓扑信息以及其他相关信息。
下面是PDB 文件的主要内容:2.1. HEADERHEADER行包含了PDB文件的标题和日期信息。
2.2. ATOMATOM行包含了蛋白质的原子坐标信息。
每个ATOM行都包含了原子序号、原子名称、残基名称、链ID、残基序号、X、Y、Z坐标、原子温度因子和元素符号等信息。
2.3. HETATMHETATM行包含了非标准氨基酸、小分子或其他非蛋白质分子的原子坐标信息。
它的格式与ATOM行相同。
2.4. TERTER行表示一个链的结束。
2.5. SEQRESSEQRES行包含了蛋白质的氨基酸序列信息。
2.6. HELIXHELIX行包含了蛋白质的α螺旋信息。
2.7. SHEETSHEET行包含了蛋白质的β折叠信息。
2.8. CONNECTCONNECT行包含了原子之间的化学键信息。
2.9. REMARKREMARK行包含了其他相关信息,如实验条件、结晶条件等。
3. PDB文件解析PDB文件的解析是指将PDB文件中的信息提取出来并进行分析。
PDB文件解析的主要步骤包括:3.1. 读取PDB文件使用程序语言(如Python)读取PDB文件中的每一行数据。
3.2. 提取原子坐标信息从ATOM和HETATM行中提取原子坐标信息,并将其存储在一个数组或矩阵中。
pdb文件蛋白质二级结构蛋白质的二级结构是指蛋白质中氨基酸残基之间的局部空间排列方式。
常见的蛋白质二级结构包括α-螺旋、β-折叠、无规卷曲和β-转角等。
在PDB文件中,蛋白质的二级结构信息通常通过特定的标记和符号进行表示。
常见的表示方法包括:1. HELIX(α-螺旋),这个标记表示蛋白质中的α-螺旋结构。
PDB文件中的HELIX记录提供了螺旋的起始残基序号、终止残基序号以及螺旋的类型。
2. SHEET(β-折叠),这个标记表示蛋白质中的β-折叠结构。
PDB文件中的SHEET记录提供了折叠片的起始残基序号、终止残基序号以及折叠片与其他折叠片之间的连接信息。
3. TURN(β-转角),这个标记表示蛋白质中的β-转角结构。
PDB文件中的TURN记录提供了转角的起始残基序号、终止残基序号以及转角的类型。
通过解析PDB文件中的这些标记和符号,可以获得蛋白质的二级结构信息。
这些信息对于研究蛋白质的结构和功能具有重要意义。
除了PDB文件中的标记和符号,还可以使用一些计算方法来预测蛋白质的二级结构。
常见的预测方法包括基于序列的方法和基于结构的方法。
基于序列的方法利用蛋白质的氨基酸序列来预测其二级结构,常见的算法有Gor方法和PSIPRED方法。
基于结构的方法则利用蛋白质的三维结构信息来进行预测,常见的算法有DSSP方法和STRIDE方法。
综上所述,蛋白质的二级结构可以通过解析PDB文件中的标记和符号得到,也可以通过计算方法进行预测。
这些信息对于理解蛋白质的结构和功能具有重要意义。
[转帖]PDB⽂件格式详解作为“⾓摩⼿机电⼦书专家”下⼀个版本的参考!PDB⽂件结构PDB是Palm Database的缩写,是⼀种简单格式的⼆进制⽂件,只要弄清楚了pdb⽂件的格式,就可以在PC上读写它。
1、 PDB⽂件的总体结构PDB⽂件总体上由以下三个部分组成,⽂件头(Header)记录索引(Record Entry Index)记录数据(Record Data)⽂件头⾥⾯主要包括数据库名称、属性、创建时间等⼀些信息。
记录索引有点像书本的⽬录部分,可以根据这个⽬录查到第⼏个记录在“第⼏页”(偏移量offset)。
数据部分才是真正的数据。
2、⽂件头⽂件头的结构如下:⽤UltraEdit打开⼀个PDB⽂件来进⾏实⼒分析PDB头Database Name图中涂蓝的部分为数据库名称(Database Name), 共21个字节,以空字符00 00结尾Create Time、Modified Time、Backup Time12字节Type、Creator ID8个字节Records Count2个字节⽂件头结束3、记录数据记录索引第⼀条记录的索引第n条记录索引的计算公式:78+(n-1)*8记录数据由第2条记录的索引可知,第2条记录的数据部分开始于00 00 00 80处,弄清了pdb的结构,可以⽤编写程序读写它,我⽤Delphi定义了⼀个pdb的类,//class PDBtypeTPDB=classprivateFDBName:String;FFlags:integer;FCreateTime:TDateTime;FModifiTime:TDateTime;FType1:String;FCreatorID:String;FRecordCount:integer;FIndex:array of integer;FAttrib:array of integer;FRecSize:array of integer;FData:array of String;procedure SetDBName(const Value: string);procedure SetFlags(const Value: integer);procedure SetCreateTime(const Value: TDateTime);procedure SetModifiTime(const Value: TDateTime);procedure SetType1(const Value: String);procedure SetCreatorID(const Value: String);procedure SetRecordCount(const Value: integer);publicConstructor create(pdbfile:string);property DBName: string read FDBName write SetDBName;property Flags:integer read FFlags write SetFlags;property CreateTime:TDateTime read FCreateTime write SetCreateTime; property ModifiTime:TDateTime read FModifiTime write SetModifiTime; property Type1:String read FType1 write SetType1;property CreatorID:String read FCreatorID write SetCreatorID;property RecordCount:integer read FRecordCount write SetRecordCount; function GetRecords(i:integer):string;end;接下来可以⽤⼀个⽂件流TFileStream读写。
pdb数据库名词解释
PDB(Protein Data Bank) 是一个蛋白质数据库,它包含了世界上大部分已知蛋白质的三维结构数据。
这些数据是由 X 光晶体衍射、NMR 等技术手段获得的,包括蛋白质分子的原子坐标、空间结构等信息。
PDB 数据库是一个关键性的资源,对于结构生物学、药物设计等领域具有重要的意义。
PDB 数据库所使用的文件格式称为“.pdb”,是一种二进制文件格式。
在 PDB 文件中,蛋白质分子的原子坐标、空间结构等信息被存储在文件的头部信息部分,而其它相关信息,如蛋白质序列、注释等则存储在文件的数据部分。
PDB 数据库的用法有多种,用户可以根据自身需求使用 PDB 数据库中的数据,例如通过查询蛋白质序列、结构信息等方式来研究蛋白质分子的结构与功能。
此外,PDB 数据库还支持用户自定义注释、模型等操作,用户可以通过这些操作来提高自己的研究水平。
需要注意的是,PDB 数据库中的数据一般是收费的,但也有一些免费的数据可以使用。
同时,由于 PDB 数据库中的数据量庞大,用户需要根据自己的需求来有选择地使用,以免浪费不必要的资源。