De novo 转录组揭示真菌与微生物的共生机制
- 格式:pdf
- 大小:6.66 MB
- 文档页数:1


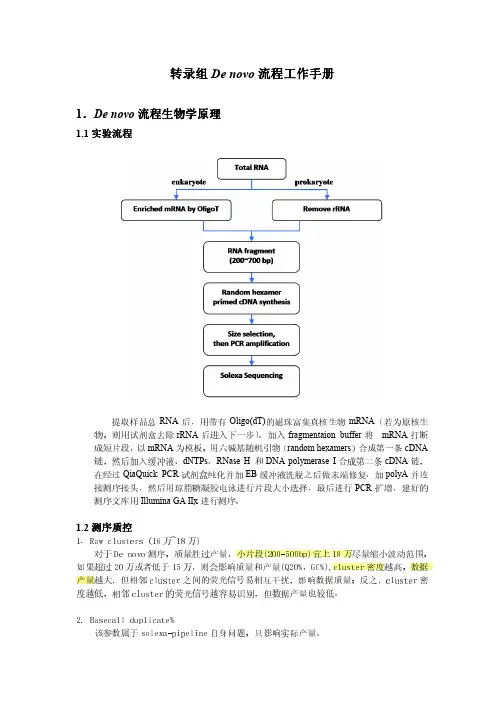

2)DNA的质量监测通常有两个方法:首先OD260/OD280比值应该在1.8左右(1.7-1.9),否则意味着DNA样品中存在大量的蛋白质或RNA污染。
其次,琼脂糖电泳分析时应主要以超螺旋条带为主。
最多不超过三条带(分别为超螺旋DNA,线性化DNA和环状DNA)。
否则意味质粒DNA的质量不高,应该重新制备。
2.限制性内切酶的活性1)限制性内切酶一般需要低温保存,而且反复的升降温过程对酶活性的损害很明显。
因而为了确保在有效期内的限制性内切酶不会失活,限制性内切酶的日常保存和使用应当很小。
2)建议购买具有保温功能的冻存盒保存限制性内切酶(-20度),而且取用限制性内切酶时,也应该使用具有保温功能的冻存盒,尽量防止酶的温度反复出现大的波动。
3.限制性内切酶的用量1)限制性内切酶的单位定义通常为:在合适的温度下,完全消化1ugDNA底物所需的酶量定义为一个单位。
2)在这个单位定义中,有几个不确定因素:首先是底物,不同的酶单位定义是选择的底物可能不同(常用的几个底物DNA包括:Lambda DNA ,AD2 DNA 和一些质粒DNA);第二个不确定因素是限制性内切酶在底物DNA上的酶切位点的个数。
由于单位定义中要求完全消化,因而底物上某个酶的酶切位点的个数的多少,就直接影响了该酶的单位定义。
3)因而,在进行酶切时,用1ul酶(一般10IU/ul)消化1ugDNA的通常做法是很不科学的,这也导致在实际工作中,大家要进行多次预实验才能确定最合适酶切条件。
4)以前,我推荐了一个在线的双酶切设计软件,double digestion designer, 可以精确地计算酶切时的限制性内切酶的用量。
使用中,能够注意到,用来进行双酶切的两个酶的用量有时竟然相差近20倍(EcoRI + NheI),而且发现,小片段PCR产物(100-500bp)进行酶切时,需要的酶量比质粒DNA酶切时用量多10倍以上。
5)该软件目前可以免费使用,用户名和密码都是test。

高通量测序基础知识汇总一代测序技术:即传统的Sanger测序法,Sanger法是根据核苷酸在待定序列模板上的引物点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以A、T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,通过检测得到DNA碱基序列。
二代测序技术:next generation sequencing(NGS)又称为高通量测序技术,与传统测序相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定,从而使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序(Deep sequencing)。
NGS主要的平台有Roche(454 & 454+),Illumina(HiSeq 2000/2500、GA IIx、MiSeq),ABI SOLiD等。
基因:Gene,是遗传的物质基础,是DNA或RNA分子上具有遗传信息的特定核苷酸序列。
基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状。
DNA:Deoxyribonucleic acid,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱基、脱氧核糖、磷酸。
脱氧核糖核酸通过3',5'-磷酸二酯键按一定的顺序彼此相连构成长链,即DNA链,DNA链上特定的核苷酸序列包含有生物的遗传信息,是绝大部分生物遗传信息的载体。
RNA:Ribonucleic Acid,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成。


微生物基因组学研究中的数据分析方法与技巧微生物基因组学是研究微生物种类和功能的学科,通过研究微生物的基因组可以了解它们的生物学特性和在环境中的角色。
而对于微生物基因组学的研究,数据分析方法和技巧是至关重要的。
本文将介绍微生物基因组学研究中常用的数据分析方法和技巧。
1.序列比对和组装技术在微生物基因组学研究中,首先要对微生物的基因组进行测序。
常用的测序技术包括Sanger测序、第二代测序(如Illumina测序)和第三代测序(如PacBio测序)。
得到基因组序列后,需要进行序列比对和组装。
序列比对是将测序获得的短序列与参考序列进行比对,以确定序列的准确位置和变异信息。
比对可以使用常见的比对工具如Bowtie2、BWA和BLAST等。
组装是将测序获得的短序列拼接成长的连续序列,以获取完整的基因组序列。
组装方法包括de novo组装和参考基因组组装。
de novo组装是从头开始组装,不需要参考序列,而参考基因组组装则是基于已有的参考序列进行组装。
2.基因预测和注释基因预测是确定基因组序列中存在的基因的位置和功能。
实现基因预测的常用工具包括Glimmer、Prodigal和GeneMark等。
通过这些工具可以预测基因的开放阅读框(ORF)和编码的蛋白质序列。
基因注释是对预测的基因进行功能描述和分类。
注释可以使用多种数据库和工具进行,如NCBI的NR和NT数据库、UniProt数据库和KEGG数据库等。
这些数据库可以提供关于基因功能、跨物种比较和代谢通路等信息。
3.基因表达分析基因表达分析是研究基因在不同条件下的表达水平和变化趋势。
常用的基因表达分析方法包括差异表达分析和聚类分析。
差异表达分析用于比较两个或多个样品(如野生型和突变型)中基因的表达差异。
常见的差异表达分析方法包括DESeq2、edgeR和limma等。
聚类分析用于将样品按照基因表达模式进行分类和分组。
常见的聚类分析方法包括层次聚类、K均值聚类和PCA等。


高通量测序常用名词汇总技术支持Q20值是指的测序过程碱基识别(Base Calling)过程中,对所识别的碱基给出的错误概率. 如果质量值是Q20,则错误识别的概率是1%,即错误率1%,或者正确率是99%;如果质量值是Q30,则错误识别的概率是0.1%,即错误率0.1%,或者正确率是99.9%;如果质量值是Q40,则错误识别的概率是0.01%,即错误率0.01%,或者正确率是99.99%;你发现规律没有,Q“N”0的质量值,就是正确率有N个9的百分比,这样就非常容易记忆了.基因高通量测序中,每测一个碱基会给出一个相应的质量值,这个质量值是衡量测序准确度的。
碱基的质量值13,错误率为5%,20的错误率为1%,30的错误率为0.1%。
行业中Q20与Q30则表示质量值≧20或30的碱基所占百分比。
例如一共测了1G的数据量,其中有0.9G的碱基质量值大于或等于20,那么Q20则为90%。
Q20值是指的测序过程碱基识别(Base Calling)过程中,对所识别的碱基给出的错误概率。
质量值是Q20,则错误识别的概率是1%,即错误率1%,或者正确率是99%;质量值是Q30,则错误识别的概率是0.1%,即错误率0.1%,或者正确率是99.9%;质量值是Q40,则错误识别的概率是0.01%,即错误率0.01%,或者正确率是99.99%;一代测序技术:即传统的Sanger测序法,Sanger法是根据核苷酸在待定序列模板上的引物点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以A、T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止,使反应得到一组长几百至几千碱基的链终止产物。

利用de novo测序分析蚕豆瓣酱醅微生物多样性周评平;李治华;董玲;赵驰;朱永清【期刊名称】《西南农业学报》【年(卷),期】2018(031)005【摘要】[目的]分析蚕豆瓣醅微生物多样性并比较与过去采用16S、ITS等扩增子测序的差异.[方法]利用de novo测序方法研究微生物菌群多样性.[结果]嗜盐四联球菌(Tetragenococcus halophilus)是蚕豆瓣酱醅的优势微生物,相对丰度占一半以上(mOTUs方法为64.26%,Metaphlan2方法为50.80%),Metaphlan2方法发现破布子乳酸菌(Lactobacillus pobuzihii)也是优势菌株,其相对丰度为17.60%,另外,存在大量未分离到的微生物有待进一步研究.[结论]该方法能分析到蚕豆瓣醅微生物中种或菌株的水平,研究结果不仅为后续深入研究提供了技术支撑,而且对郫县豆瓣的微生物安全性评价提供了有效的方法.【总页数】3页(P1055-1057)【作者】周评平;李治华;董玲;赵驰;朱永清【作者单位】四川省农业科学院,四川成都610066;四川省农业科学院农产品加工研究所,四川成都610066;四川省农业科学院农产品加工研究所,四川成都610066;四川省农业科学院农产品加工研究所,四川成都610066;四川省农业科学院农产品加工研究所,四川成都610066;四川省农业科学院农产品加工研究所,四川成都610066【正文语种】中文【中图分类】Q93【相关文献】1.利用de novo组装策略解析猪的基因组变异 [J], 田仕林;唐茜子;李学伟;李明洲2.一株耐受蛋清蛋白的鸡蛋腐败菌S菌株的分离鉴定及其De novo测序分析 [J], 张璟;陈倩;顾丽红;阮瑶;张新帅;郭爱玲3.应用Illumina MiSeq高通量测序技术分析巴东地区豆瓣酱中微生物多样性 [J], 赵馨馨;崔梦君;董蕴;倪慧;单春会;郭壮4.基于高通量测序分析玉米黄酒微生物多样性 [J], 李永翔;蒋海娇;郭建华5.不同来源及不同发酵阶段豆瓣酱微生物多样性的比较分析 [J], 杨帆;邓维琴;李恒;唐浪;马嫄因版权原因,仅展示原文概要,查看原文内容请购买。

转录组学微生物转录组学微生物是一门研究微生物基因表达的学科,通过对微生物转录组的分析,可以深入了解微生物在不同环境中的基因表达模式,揭示微生物的生理特性和生物功能。
本文将从转录组学的基本原理、研究方法和应用领域等方面进行介绍。
一、转录组学基本原理转录组是指一个生物体在某个时刻的所有基因的转录产物,即所有mRNA的总和。
转录组学研究的主要目标是通过高通量测序技术,对微生物的转录产物进行全面和系统地分析,以获得微生物在特定环境中的基因表达谱。
转录组学的研究基于以下两个基本原理:1. 基因表达的可变性:微生物在不同环境中的基因表达模式会发生变化,这种变化可以通过转录组学的分析来揭示。
通过比较不同条件下的转录组数据,可以了解微生物对环境的适应机制,以及其在不同生长阶段或不同环境中的适应策略。
2. 基因调控网络:微生物的基因表达受到复杂的调控网络控制,包括转录因子、信号传导通路和代谢途径等。
转录组学可以揭示这些调控网络的结构和功能,帮助我们理解微生物的生物学过程和生物功能。
二、转录组学研究方法转录组学的研究方法主要包括以下几个步骤:1. RNA提取:从微生物样品中提取RNA,包括mRNA和非编码RNA等。
2. RNA测序:使用高通量测序技术对RNA样品进行测序,得到大量的短序列数据。
3. 数据分析:对测序数据进行质控、比对和注释等分析,得到基因表达谱和差异表达基因。
4. 功能注释:对差异表达基因进行功能注释和富集分析,了解微生物的生物学功能和代谢途径等。
5. 转录因子预测:通过分析转录因子结合位点和转录因子基因的表达数据,预测微生物的转录因子和调控网络。
三、转录组学在微生物研究中的应用转录组学在微生物研究中有广泛的应用,主要包括以下几个方面:1. 研究微生物的适应机制:通过比较不同环境下的转录组数据,可以了解微生物对环境的适应机制和适应策略。
例如,研究细菌在不同营养条件下的基因表达模式,可以揭示其对不同营养物质的利用方式和代谢途径。
真菌基因组de novo测序项目方案背景介绍从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。
利用全基因组从头测序技术,可以获得全基因组序列,从而推进该物种的研究。
全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。
一、项目概述采用全基因组鸟枪法(WGS)策略,将Illumina与PacBio相结合,构建该真菌基因组的框架图。
二、技术方案(一)方案1:测序平台:Illumina PE + Illumina MPIllumina测序平台的读长较短,但可以利用其通量高的特点,对基因组进行深度测序,搭配段片段PE(pair-end)文库和长片段的MP(mate-pair)文库进行测序。
技术优势:因为Illumina读长短,利用MP(mate-pair)文库测序可以在一定程度上提高拼接的质量。
测序数据量:该物种的基因组大小100Mb,建议Illumina数据覆盖300X,即30Gb以上数据。
(二)方案2:测序平台:Illumina PE + PacBio第三代测序平台PacBio具有读长较长的优势(读长10 kb),能够在序列上通过重复序列区及高GC区,从而达到更好的拼接效果;同时,利用第二代测序平台Illumina(HiSeq 2500)数据量大、成本低的优势,对小插入片段基因组文库(插入片段500 bp)进行双末端(Paired-End)测序。
技术优势:因为读长很长,所以在拼Contig时,成功率很高,可以拼出很长的Contig。
并且可以轻松跨过重复序列、高GC序列。
实际应用中,大家普遍用PacBio序列拼Contig,再用Illumina的序列来修正碱基。
测序数据量:该物种的基因组大小100Mb,建议Illumina数据覆盖100X,即10Gb以上数据;PacBio数据5~10X,即0.5~1Gb数据,也就是2~4个SMRT cell。
#流程大放送#微生物基因组Denovo测序分析知因无限一介绍微生物基因组De novo测序分析也叫微生物基因组从头测序分析,指不依赖于任何参考序列信息就可对某个微生物进行分析的测序分析技术,用生物信息学的方法进行序列拼接获得该物种的基因组序列图谱,然后进行注释等后续一系列的分析。
微生物Denovo基因组测序及分析技术可以应用于医药卫生等领域。
二技术应用领域1、基因组图谱的系统性构建例子:过去几个月,肠病毒D68令数百名美国儿童患病。
华盛顿大学的研究人员测序和分析了肠病毒D68(EV-D68)的基因组,这一成果将发表在新一期的Emerging Infectious Diseases杂志上。
(Genome Sequence of Enterovirus D68 from St. Louis, Missouri, USA)肠病毒D68(EV-D68)能在儿童中引起严重的呼吸道疾病。
其基因组序列可以“帮助人们开发更好的诊断测试,”共同作者Gregory Storch说。
“有助于解释病毒感染为什么会造成严重的疾病,以及EV-D68为什么比过去传播得更广。
”(来自于生物通的报道)2、微生物致病性和耐药性位点检测及相关基因功能研究例子:根据分泌蛋白、毒力因子、致病岛、必需基因等结果去探讨所测物种致病性和耐药性。
3、微生物的比较基因组分析,确定各个近缘微生物中的系统发育关系二基本分析流程图三可能的结果展示图示例图1 微生物基因组的功能注释示例图2 微生物基因组的系统进化关系注:以上图片和文字来自参考文献21。
六参考文献[1] Hong-Bin Shen, and Kuo-Chen Chou, "Virus-mPLoc: a fusion classifier for viral protein subcellular location prediction by incorporating multiple sites", Journal of Biomolecular Structure & Dynamics, 2010, 28: 175-86.[2]Hong-Bin Shen and Kuo-Chen Chou, "Virus-PLoc: A fusion classifier for predicting the subcellular localization of viral proteins within host and virus-infected cells.", Biopolymers. 2007, 85, 233-240.[3] Ren Zhang and Yan Lin, (2009) DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Research 37, D455-D458.[4] The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007 May 23;8(1):172.[5] The Pfam protein families database: M. Punta, P.C. Coggill, R.Y. Eberhardt, J. Mistry, J. Tate,C. Boursnell, N. Pang, K. Forslund, G. Ceric, J. Clements, A. Heger, L. Holm, E.L.L. Sonnhammer, S.R. Eddy, A. Bateman, R.D. Finn Nucleic Acids Research (2014) Database Issue 42:D222-D230.[6] Clustal W and Clustal X version 2.0.(2007 Nov 01) Bioinformatics (Oxford, England) 23 (21) :2947-8.PMID: 17846036.[7] Felsenstein, J. 2004. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle.[8] Li et al (2010). De novo assembly of human genomes with massively parallel short readsequencing. Genome Res vol. 20 (2).[9] Li et al (2008). SOAP: short oligonucleotide alignment program. Bioinformatics Vol. 24 no.5 2008.[10] A.L. Delcher, D. Harmon, S. Kasif, O. White, and S.L. Salzberg (1999) Improved microbial gene identification with GLIMMER, Nucleic Acids Research 27:23 4636-4641.[11] S. Salzberg, A. Delcher, S. Kasif, and O. White (1998) Microbial gene identification using interpolated Markov models, Nucleic Acids Research 26:2, 544-548.[12] Delcher AL, Bratke KA Powe,rs EC,et al(2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics,23(6):673-679.[13]G. Benson(1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research, Vol. 27, No. 2, pp. 573-580.[14] Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res 32 (Database issue): D277–80.[15] Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34(Database issue): D354–7.[16] Tatusov RL, Koonin EV, Lipman DJ(1997). A genomic perspective on protein families. Science. Oct 24;278(5338):631-7.[17] Tatusov RL, Fedorova ND et al.(2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics. Sep 11;4:41.[18] Magrane, M. and UniProt Consortium (2011) UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) , bar009.[19] Bard J, Winter R (2000). Gene Ontology:tool for the unification of biology. Nat Genet. 25:25-29.[20] ZODOBNOV.E.M,APWEILER.R.InterProScan—an intergration plaftorm forthe signature recognition methods in InterPro[J].Bioinform atics,2001,17(9):847-848.[21] Van den Bogert B1, Boekhorst J2, Herrmann R1, Smid EJ3, Zoetendal EG1, Kleerebezem M4. Comparative genomics analysis of Streptococcus isolates from the human small intestine reveals their adaptation to a highly dynamic ecosystem. PLoS One. 2013 Dec 30;8(12):e83418.。
动植物Denovo测序知识⼤讲解⾼通量测序的技术开起我们探索动植物基因组奥秘的步伐,提到动植物基因组测序,这就不得不提⼀个概念——de novo测序。
那么什么是de nove测序呢,它与重测序有什么区别呢?De nove测序中Read、Contig和Scaffold等⼜代表什么呢?De nove测序中为什么要建不同⼤⼩⽚段的梯度⽂库?基因注释⼜是注释哪些内容?各位客官别急,且听⼩编给您细细讲来。
1De novo测序概念De novo是⼀个拉丁⽂,代表从头开始的意思,⽽de nove测序则是指在不需要任何参考序列的情况下对某⼀物种进⾏基因组测序,然后将测得的序列进⾏拼接、组装,从⽽绘制该物种的全基因组序列图谱。
由于⾼通量测序长度的限制,⽬前测序策略是先将基因组打断⼩的⽚段,然后再对测出序列⽚段进⾏拼接,最终得到物种的序列图谱如图1所⽰。
图1 ⾼通量测序模式图2De novo测序与重测序区别重测序概念:重测序是全基因组重新测序的简称,是指是对已知基因组序列的物种进⾏不同个体的基因组测序,并在此基础上对个体或群体进⾏差异性分析。
从概念上来看两者的区别在于de nove测序是对没有参考基因组的物种进⾏测序,⽽重测序是对已有基因组的物种进⾏测序,这只是它们区别很⼩的⼀部分。
从原理上来看de nove测序和重测序最根本的区别在于de nove测序需要对测序得到的Reads进⾏拼接组装,⽽重测序得到的数据则是没有组装的短的Reads序列。
值得注意的是,随着测序成本的降低以及组装算法的改进,de nove测序成本越来越低,⽬前来说de nove测序不只对于没有参考基因组物种进⾏测序,还可以对⼀些特有的亚种、品种以及变种等进⾏测序。
3Reads Conting Scaffold概念Reads:即我们通常说的读长的意思,它是指⾼通量测序平台直接产⽣的DNA序列。
Contig:是指Reads基于Overlap关系,拼接获得的长的序列;Scaffold:是指将获得的Contig根据⼤⽚段⽂库的Pair-end关系,将Contig进⼀步组装成更长的序列;关于三者之间的关系如图2所⽰,注意的是Contig是⽆Gap的连续的DNA序列,⽽Scaffold是存在Gap的DNA序列。
基因组denovo深度基因组de novo深度是一种重要的研究方法,可以帮助我们理解生物体的遗传信息。
在这篇文章中,我将以人类的视角来描述这一方法的原理和应用。
让我们来看看什么是基因组de novo深度。
简单来说,它是一种通过测序技术从头开始组装一个生物体的基因组的方法。
与传统的测序方法不同,de novo深度测序可以直接获得一个生物体的全基因组信息,而不需参考已有的相关序列。
那么,为什么我们需要基因组de novo深度呢?这是因为在许多研究中,我们需要了解一个生物体的完整基因组信息,尤其是对于那些没有已知参考基因组的物种来说。
通过de novo深度测序,我们可以获得这些物种的全基因组序列,从而更好地理解它们的遗传特性和进化历史。
在进行基因组de novo深度测序时,首先需要将生物体的DNA提取出来,并进行高通量测序。
然后,利用生物信息学的方法将这些测序数据进行组装,得到一个生物体的基因组序列。
这个过程中,需要借助大量的计算资源和算法,以及对基因组结构和功能的理解。
基因组de novo深度的应用非常广泛。
例如,它可以帮助我们研究物种的进化关系、基因组结构的变异以及基因与表型之间的关系。
同时,它也可以用于研究人类疾病的遗传基础,例如发育异常、遗传疾病和癌症等。
尽管基因组de novo深度是一项复杂的技术,但它为我们揭示了生命的奥秘提供了重要的工具。
通过这种方法,我们可以更好地理解生物体的基因组,为生物学研究和医学应用提供更多的可能性。
基因组de novo深度是一种重要的研究方法,它可以帮助我们获得生物体的完整基因组信息,从而更好地了解生物体的遗传特性和进化历史。
它的应用范围广泛,可以用于研究进化、遗传疾病等领域。
通过不断地改进和发展,基因组de novo深度将为我们揭示生命的奥秘提供更多的突破。
专利名称:采用De novo转录组分析鉴定隐甲藻中与DHA生物合成相关的脂肪酸去饱和酶基因的方法
专利类型:发明专利
发明人:张卫文,陈磊,裴广胜,刘璐
申请号:CN201710105914.8
申请日:20170224
公开号:CN106916891A
公开日:
20170704
专利内容由知识产权出版社提供
摘要:本发明公开了一种通过De novo转录组测序,转录本拼接和差异表达分析,鉴定蔻氏隐甲藻中与二十二碳六烯酸(DHA)生物合成相关脂肪酸去饱和酶基因的方法。
具体步骤包括:(1)对隐甲藻进行DHA发酵培养;(2)收集不同生长和DHA积累阶段的菌体并进行RNA提取与转录组Illumina深度测序;(3)测序数据的分析与预处理;(4)对拼接后的转录组进行功能注释与相关性统计分析,鉴定发现隐甲藻中与DHA生物合成相关的脂肪酸去饱和酶,并获得其基因全长序列;(5)通过qRT‑PCR分析技术,对隐甲藻中与DHA生物合成相关的脂肪酸去饱和酶的基因进行表达验证。
发明为研究隐甲藻DHA的生物合成机制以及提高隐甲藻中DHA合能力成提供重要的方法和知识基础。
申请人:昆明藻能生物科技有限公司
地址:650108 云南省昆明市科发路139号云南省大学科技园云南留学人员创业园二期A3栋5楼505-3号
国籍:CN
代理机构:昆明今威专利商标代理有限公司
代理人:赛晓刚
更多信息请下载全文后查看。
然而,利用以上研究数据,另有文章发表:
NGS项目文章
猪苓,非褶菌目多孔菌科树花属,是一种药用真菌;
蜜环菌,伞菌目蜜环菌属,夏秋季常寄生于树丛根部,是一种根腐菌。
猪苓的生长需要蜜环菌提供营养,二者之间存在共生关系。
在共生关系建立的过程中,蜜环菌的菌索会吸附并侵染猪苓的菌核,
而猪苓则会激发自身防御机制,如菌核细胞壁不规则地增厚以抵抗蜜环菌进一步侵染。
这个抵抗侵染过程的分子机制是怎样的?
2015年10月,猪苓对蜜环菌的防御机制研究成果发表在Scientific Reports上,
该研究由中国医学科学院药用植物研究所郭顺星教授课题组负责,
其中De novo
转录组测序工作由诺禾致源完成。
研究内容与结果
首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们
De novo 转录组
揭示猪苓对蜜环菌的防御机制
3. 差异基因功能分析
利用DESeq软件进行CT和CK的差异基因筛选,得到10,933个DEG,包括
8,780个上调基因和2,213个下调基因(图2)。
表达上调的差异基因GO富集分析发现:在molecular function部分,富集
最明显的是苏氨酸型肽链内切酶活性、肽酶活性以及特异性DNA结合等
term;在cellular component部分,富集显著的是蛋白酶复合体、膜组成
等term;而在biological process部分,蛋白质折叠、三羧酸循环以及染色
质组装等term下显著富集。
而下调的DEG被富集到对有机物反应、细胞组
分合成等功能term,这预示着猪苓受到侵染时会限制自身的生长与繁殖。
对DEG进行KEGG富集分析发现:精氨酸和脯氨酸代谢通路富集显著;免疫
相关基因精氨酸酶(ARG)显著上调,推测其与猪苓抵抗蜜环菌侵染相关。
而在核糖体合成途径通路中,含有11个下调基因,这与GO富集中发现下调
基因在生物学过程相关term富集的结果是一致的,从而推测猪苓受到侵染
会减弱自身的生物学过程以保存能量。
2. 转录本功能注释
对拼接得到的转录本进行功
能注释,采用Nr, Nt, Swiss
-Prot, KEGG, GO, KOG 和
Pfam 七大数据库。
并对
unigene进行GO和KOG功
能分类(图1和图2)。
1. 转录本拼接
四个文库测序下机的原始
数据进行质控,将各样本
的clean reads进行混合拼
接,得到38444条
unigene,作为参考序列用
于后续分析。
4. qPCR验证差异表达基因
选取13个DEG进行实时荧光定量PCR,检测结果与RNA-seq一致。
结论与讨论
本研究首次通过RNA-seq研究蜜环菌侵染和未侵染的猪苓样本,筛选出差异表达基因10933个;
通过GO富集和KEGG富集分析,鉴定到了可能与抵御反应相关的若干差异基因。
其中,且热休克蛋白(Hsps)基因、抗氧化防御相关基因、凝集素基因、致病相关蛋白(TLP)基因、
次级代谢相关基因、细胞壁水解与融合相关基因、PDR家族基因、WD40蛋白基因等
在蜜环菌侵染的猪苓样本中呈现表达上调,均可能与抵御反应相关。
图3 转录数据的SSR分析
图2 对差异基因进行筛选
图1 unigene的GO功能分类
郭顺星教授课题组继续利用本次RNA-seq的数据结
果进行SSR分析,丰富了药用猪苓的因地域而异的
遗传多态性,并为猪苓群体遗传和行为生态学研究
提供了依据。
阅读原文>>
阅读原文>>
Scientific Reports。