ClustalX 软件使用简易教程
- 格式:doc
- 大小:35.50 KB
- 文档页数:12


Clustalx的中文使用说明书
生物
用ClustalX做多序列比对分析图示
1、打开程序如下图所示:
2、Load Sequnce, 载入序列如下图所示:
3、选择序列文件,FASTA格式的如下图所示:
4、用文本编辑器察看FASTA序列文件内容,这里用的是记事本,推荐用EditPlus或者Ultraedit 如下图所示:
5、序列Load进去之后如下图所示:
6、Do Complete Alignment, 通常情况下直接选这个即可,无须修改比对参数如下图所示:
7、点Do Complete Alignment之后弹出的文件对话框,.dnd的是输出的指导树文件,.aln 的是序列比对结果,它们都是纯文本文件如下图所示:
点“ALIGN”之后开始等待,如果序列不多,很快就可以算完,如果数据很多,可能要等一段时间,这时候可以用眼睛盯着ClustalX的状态栏,那里会有程序运行状态和现在正在比对那两条序列的提示信息,看看可以消磨时间。
8、比对结束之后,我们可以看到这个结果如下图所示:
9、这时候我们可以发现ClustalX已经生成了.dnd和.aln两个文件,仍然用文本编辑器打开来看,这时.aln文件,这个文件可以用Mega2做进一步的bootstrap进化树分析如下图所示:
10、这是.dnd文件(指导树) 如下图所示:
11、可以用Treeview打开dnd文件,看上去就像这样子如下图所示
图3-15 ClustalX所识别的文件输入格式。


【软件使⽤】clustalX2使⽤以及相关的问题Clustalx的操作第⼀步:输⼊序列⽂件。
第⼆步:设定⽐对的⼀些参数。
参数设定窗⼝。
第三步:开始序列⽐对。
第四步:⽐对完成,选择保存结果⽂件的格式相关问题CLUSTALX-是CLUSTAL多重序列⽐对程序的Windows版本。
Clustal X为进⾏多重序列和轮廓⽐对和分析结果提供⼀个整体的环境。
这⾥总结⼀下在使⽤做序列⽐对过程中⼀些常见的问题~1,从⽹上看来说CLUSTALX软件使⽤的时候,开始要输⼊FASTA格式准备的DNA序列test.seq⽂件。
请问这种⽂件怎么⽣成啊?记事本?其实没有规定的⽂件名,不⼀定是叫test.seq,任何你想要的都可以,除了中⽂名(下⾯会说明)。
主要⾥⾯的内容是正确的格式就⾏了~。
Clustal⽀持的格式有多种,如NBRF/PIR, EMBL/SWISSPROT, Pearson (Fasta), Clustal (*.aln), GCG/MSF (Pileup), GCG9/RSF and GDE 等2,什么使⽤时,总是导⼊不了序列,出现 ERROR:Can not open output file??这个问题挺多⼈碰到过的~这是因为你导⼊的⽂件的路径包含有中⽂名。
把⽂件放在其它地⽅看看。
最好路径也不要有空格了。
当然前提是你的格式要正确了。
这个问题同样适⽤于ClustalW或其它dos类的软件3,ClustalX⽐对后的结果中.aln⽂件是什么?这个是序列⽐对结果的⽂件。
关于这部分,你可以看4,ClustalX⽐对后的结果中.dnd⽂件是什么?。
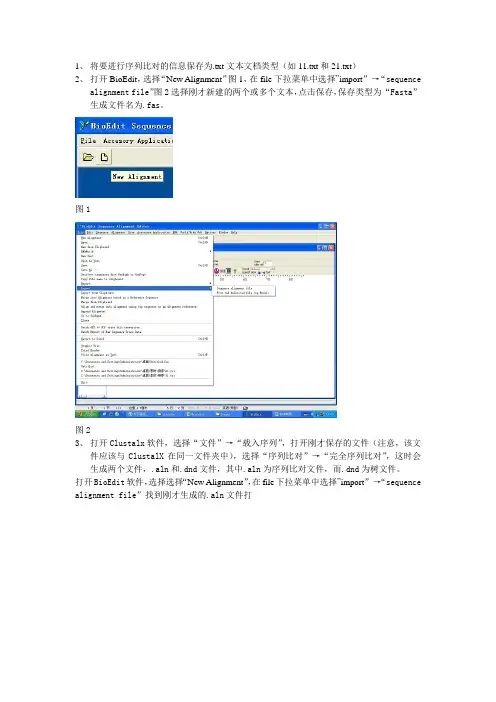
1、将要进行序列比对的信息保存为.txt文本文档类型(如11.txt和21.txt)
2、打开BioEdit,选择“New Alignment”图1,在file下拉菜单中选择”import”→“sequence
alignment file”图2选择刚才新建的两个或多个文本,点击保存,保存类型为“Fasta”
生成文件名为.fas。
图1
图2
3、打开Clustalx软件,选择“文件”→“载入序列”,打开刚才保存的文件(注意,该文
件应该与ClustalX在同一文件夹中),选择“序列比对”→“完全序列比对”,这时会生成两个文件,.aln和.dnd文件,其中.aln为序列比对文件,而.dnd为树文件。
打开BioEdit软件,选择选择“New Alignment”,在file下拉菜单中选择”import”→“sequence alignment file”找到刚才生成的.aln文件打。

如何用MEGA5.0和Clustalx1.83构建进化树MEGA是一个关于序列分析以及比较统计的工具包,从3.1版本到后来的4.0版本一直都广为大家熟悉,现在推出了Mega5.0版本。
功能比以前多有改进。
现主要介绍使用Mega 5.0构建系统进化树的方法。
供大家参考。
用MEGA构建进化树有以下步骤:1、测序:将克隆扩增测序得到的16S rDNA序列进行测序。
2、NCBI上做Blast/blast/Blast.cgi找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后寻找相似性最高的细菌,通常把该属的序列(Fasta格式文件)下载下来,或点击GenBank登录号,复制FSA TA 格式,整合在一个*.txt文档中(单独建立一个文件夹存放,后面的很多文件会自动装入该文件夹),如>XXXXAGGCTTAACACA TGCAAGTCGAGCGGAGCGAGGGTGCTTGCACCTTAGCTTAGCGGCG GACGGGTGAGTAA TGCTTAGGAA TCTGCCTA TTAGTGGGGGACAACA TTCCGAAAGGA A TGCTAA TACCGCA TACGCCCTACGGGGGAAAGCAGGGGA TCTTCGGACCTTGCGCTAA TAGA TGAGCCTAAGTCGGA TTAGCTAGTTGGTGGG>gi|289469964|gb|GU388381.1| Acinetobacter tandoii strain DSM 14970 16S ribosomal RNA gene, partial sequenceACTTAGCGGCGGACGGGTGAGTAA TGCTTAGGAA TCTGCCTA TTAGTGGGGGACAACA TTCCGAAAGGGA TGCTAA TACCGCA TACGCCCTACGGGGGAAAGCAGGGGA TCTTCGG ACCTTGCGCTAA TAGA TGAGCCTAAGTCGGA TTAGCTAGTTGGTGGGGTAAAGGCCTAC CAAGGCGACGA TCTGTAGCGGGTCTGAGAGGA TGA………………………….参考序列选择注意事项:1、不选非培养(unclutured)微生物为参比;2、不选未定分类地位的微生物,最相近的仅作参考;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列。

C l u s t a l x多重序列比对图解教程(B y R a i n d y) 本帖首发于Raindy'blog软件简介:CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
ClustalX为进行多重序列和轮廓比对和分析结果提供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征。
窗口上面的下拉菜单可让你选择传统多重比对和轮廓比对需要的所有选项。
主要功能:你可以剪切、粘贴序列以更改比对的顺序;你可以选择序列子集进行比对;你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx1.81版链接地址:ist&ID=7435(请完整复制)应用:Clustalx比对结果是构建系统发育树的前提实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方的菜单栏选择“File”-“LoadSequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是否替换现有序列(Replaceexistingsequences),根据具体情形选择操作。
图1图22.编辑序列:对标尺(Ruler)上方的序列进行编辑操作,主要有Cutsequences(剪切序列)、Pastesequences(粘贴)、SelectAllsequences(选定所有序列),ClearsequenceSelection(清除序列选定)、Searchforstring(搜索字串)、RemoveAllgaps(移除序列空位)、RemoveGap-OnlyColumns(仅移除选定序列的空位)图33.参数设置:可以根据分析要求设置相对的比对参数。


Clustalx 多重序列比对图解教程(By Raindy)本帖首发于Raindy'blog,转载请保留作者信息,谢谢!欢迎有写生物学软件专长的战友,加入生信教程写作群:13559330,接头暗号:你所擅长的生物学软件名称软件简介:CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
Clustal X为进行多重序列和轮廓比对和分析结果提供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征。
窗口上面的下拉菜单可让你选择传统多重比对和轮廓比对需要的所有选项。
主要功能:你可以剪切、粘贴序列以更改比对的顺序;你可以选择序列子集进行比对;你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx 1.81版链接地址:/index.php?Go=Show:ist&ID=7435 (请完整复制)应用:Clustalx比对结果是构建系统发育树的前提实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方的菜单栏选择“File”-“Load Sequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是否替换现有序列(Replace existing sequences),根据具体情形选择操作。
图1图22.编辑序列:对标尺(Ruler)上方的序列进行编辑操作,主要有Cut sequences(剪切序列)、Paste sequences(粘贴)、Select All sequences(选定所有序列),Clear sequence Selection(清除序列选定)、Search for string(搜索字串)、Remove All gaps(移除序列空位)、Remove Gap-Only Columns(仅移除选定序列的空位)图33.参数设置:可以根据分析要求设置相对的比对参数。

序列比对之Clustalx与Clustalw使用指南这几天实验需要做多序列比对,很久不做了,一时之间不知道如何使用clustal这个工具了。
在网上搜集了一些资料,做个整理,总结了Clustalx和Clustalw的使用,省得以后久不使用又生疏了,又要去整理了,在此分享给大家,希望有所帮助。
1.先提供下载地址:官方下载地址:/download/current/Clustalx、Clustalw的各种最新版本都能下载到,包括linux、Win、Mac...2.原理:序列同源性分析:是将待研究序列加入到一组与之同源,但来自不同物种的序列中进行多序列同时比较,以确定该序列与其它序列间的同源性大小。
这是理论分析方法中最关键的一步。
完成这一工作必须使用多序列比较算法。
常用的程序包有CLUSTAL等;Clustal是一个单机版的基于渐进比对的多序列比对工具,由Higgins D.G.等开发。
有应用于多种操作系统平台的版本,包括linux版,DOS版的clustlw,clustalx等。
CLUSTAL是一种渐进的比对方法,先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树,对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止。
3.操作3.1 Clustalx的操作第一步:输入序列文件。
第二步:设定比对的一些参数。
参数设定窗口。
第三步:开始序列比对。
第四步:比对完成,选择保存结果文件的格式--------------------------------------------3.2 Clustalw的使用(一)第一步:按屏幕提示选择1,输入序列文件注意:请把你需要比对的多条序列合并为一条,放在一个文件中第二步:选择保存文件的形式,按提示选择,你会的!第三步:参数设置好后,按Enter返回主界面,开始比对!EBI提供的在线clustalw服务/clustalw/可以在这里得到更多关于clustal的帮助:http://www-igbmc.u-strasbg.fr/BioInfo/ClustalX/Top.html。

clustalx多序列比对结果建树
在ClustalX中进行多序列比对后,可以使用其提供的建树工
具将比对结果转化为树状结构。
以下是使用ClustalX建树的
步骤:
1. 打开ClustalX软件,点击"File",选择"Open Alignment",选择之前进行多序列比对的文件。
2. 点击"Tree",选择"Build Tree",进入建树参数设置界面。
3. 在参数设置界面中,可以选择不同的建树方法,如
Neighbor-Joining、UPGMA等。
可以根据需要选择合适的方法。
4. 设置完参数后,点击"OK"开始建树过程。
5. 建树完成后,可以在软件界面的右侧窗口中查看树状结构。
可以通过点击树状图上的节点来展开或折叠子树。
需要注意的是,ClustalX建树功能只是提供了一种简单的可视
化方法,用于初步了解序列的进化关系。
如果需要更加精确的进化关系推断,可以考虑使用其他更专业的建树软件如PhyML、PAUP*等。
利用clustalx 2.1对蛋白进行多序列比对目录1. 方法介绍1.1概念1.2理论基础1.3任务1.4目的2研究内容3. 工具3.1 clustalx简介3.2 clustalx 后台运作流程3.3 clustalx的下载3.4 clustalx菜单设置4.操作步骤4.1获取目标序列4.2执行比对4.3 treeview软件制作进化树5. 结果分析正文1. 方法介绍:多序列比对1.1 概念:多序列比对即通过多个核苷酸或氨基酸的序列进行比较,确定序列之间可能由于功能、结构或进化上的关联而形成的相似片段。
1.2 理论基础:1)生物学一个最基本的假设是地球上所有物种都有共同的祖先,从这个祖先开始以树状形式发展,通常称为生命之树。
2)基于序列比对的同源即具有共同祖先。
同源序列一般相似;相似可以用百分比来描述。
序列不一定是同源的,相似序列在进化上具有趋同性。
序列决定结构,结构决定功能。
3)现有的基因、蛋白质等携带生物学信息、具有生物学功能的分子都是由原有的分子演化而来;现有的基因及其他核酸序列,都是由已经存在的基因或其他序列经过复制、转移、合并、删减等方式形成的;不同物种的基因、蛋白质在结构、序列上的相似性与其进化上亲缘关系密切相关。
1.3 任务:发现序列之间的相似性,找出序列之间共同的区域,辨别序列之间的差异。
1.4 目的:通过“相似序列→相似的结构→相似的功能“来判别序列之间的同源性,进而推测序列之间的进化关系。
2. 研究内容:通过对人类、家鼠、大鼠和鸡体内BMP-2(bone morphogeneticprotein 2)即骨形态发生蛋白2的多序列比对得到的dnd结果文件来揭示在四种生物中的该蛋白的同源性。
3. 工具:clustalx 2.13.1 clustalx简介:Clustal是用来对核酸与蛋白序列进行多序列比对的软件,可以用来发现特征序列,进行蛋白分类,证明序列间的同源性,帮助预测新序列二级结构与三级结构,确定PCR引物,以及在分子进化分析方面均有很大帮助。
ClustalX Help可以在下列地址得到 ClustalX 的最新版本:ftp://ftp-igbmc.u-strasbg.fr/pub/ClustalX/General help for CLUSTAL X (1.8)Clustal X 是一个windows 界面多序列对比程序。
可以用剪切和粘贴的方法改变对比的顺序;可以在比对中选择较小的区域重新比对,并将比对的结果插回到原来的比对结果中。
能够进行比对质量评定,低分片断和多余的残基将突出显示。
序列输入序列和轮廓(已经存在的比对)利用菜单文件输入,所有的序列必须放到一个文件中,7种序列可以被自动识别: NBRF/PIR, EMBL/SWISSPROT, Pearson (Fasta), Clustal (*.aln), GCG/MSF (Pileup), 除用于表示间隙的"-" 例外 ("." in MSF/RSF),所有的非字母字符将被忽略。
序列和轮廓比对Clustal X 有两种比对格式: 多重比对格式和轮廓比对格式。
做一系列序列的多重比对时要保证选择多重比对模式,然后展示单一序列数据。
比对菜单既可以产生比对的指导树又可根据指导树进行比对,还可以做全多重比对。
在轮廓比对模式下,出现两个序列数据区,允许对两个比对结果进行比对。
轮廓允许添加新序列到旧的比对中,或者应用二级结构指导比对进程。
旧比对中的间隙用 "-"表示。
轮廓可以用以下任何一种格式输入,只有用 "-" (or "." for MSF/RSF) 代表每一个间隙位置。
在轮廓比对状态下, "Lock Scroll"按钮 is displayed which allows you to scroll the two profiles together using a single scroll bar. When the Lock Scroll is turned off, the two profiles can be scrolled independently.进化树进化树可以从旧的比对或新比对中产生。
实验三:多条序列比对——Clustalx(一)ClustalXClustal是一种利用渐近法(progressive alignment)进行多条序列比对的软件。
即从多条序列中最相似(距离最近)的两条序列开始比对,按照各个序列在进化树上的位置,由近及远的将其它序列依次加入到最终的比对结果。
(Figure 3.1)/1.安装clustalx程序。
双击安装clustalx-2.0.12-win.msi.exe文件到自己的电脑上。
也可从/download/current/下载,列表中的倒数第二个文件。
clustalx-2.0.12-win.msiFigure 3.1 clustal 算法2.准备要比对的序列请查找至少存在于5个物种中的同源序列(核酸或蛋白质皆可),并保存为fasta格式,存为文本文件(所有的序列请粘贴到同一个文本文件中)。
选择NM、XM或NP打头的序列,不要选择NC或NW打头的序列,那是全基因组序列。
做法可参照邮箱中的preparations for practice3.doc文件。
3.打开clustalX程序开始菜单-程序-clustalX2- clustalX24.载入序列点最上方的File菜单,选择Load Sequence-选择你刚保存的序列文件,点打开。
在左侧窗口里是fasta格式序列的标识号,取自序列第一行“>”后的字符。
(Figure 3.2) 注意:ClustalX程序无法识别汉字,无法识别带空位的文件夹名,如 my document。
各位同学保存的序列文件不要保存在桌面上或带汉字的文件夹中,推荐保存在D盘根目录下。
常见文件打开错误原因:1.序列格式有问题,非正确的fasta格式。
2.文件中有序列重复粘贴。
TIPS: 想要方便识别序列所属物种,可在每条序列“>”后输入物种名,加空位即可。
EXAMPLE:原格式:>gi|262050536|ref|NM_002218.4| Homo sapiens inter-alpha (globulin) inhibitor H4 (plasma Kallikrein-sensitive glycoprotein) (ITIH4), transcript variant 1, mRNA改为:>human gi|262050536|ref|NM_002218.4| Homo sapiens inter-alpha (globulin) inhibitor H4 (plasma Kallikrein-sensitive glycoprotein) (ITIH4), transcript variant 1, mRNAFigure 3.2 载入序列5.比对参数的选择可以对两条序列比对的参数和多条序列比对的参数进行设置。
本帖首发于Raindy'blog,
软件简介:
CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
Clustal X为进行供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征传统多重比对和轮廓比对需要的所有选项。
主要功能:
你可以剪切、粘贴序列以更改比对的顺序;
你可以选择序列子集进行比对;
你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;
可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83
PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx 1.81版链接地址:/index.php?Go=Show::List&ID=7435(请
应用:Clustalx比对结果是构建系统发育树的前提
实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例
流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果
1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方Sequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是sequences),根据具体情形选择操作。