制剂分析方法的建立和验证
- 格式:ppt
- 大小:2.28 MB
- 文档页数:109
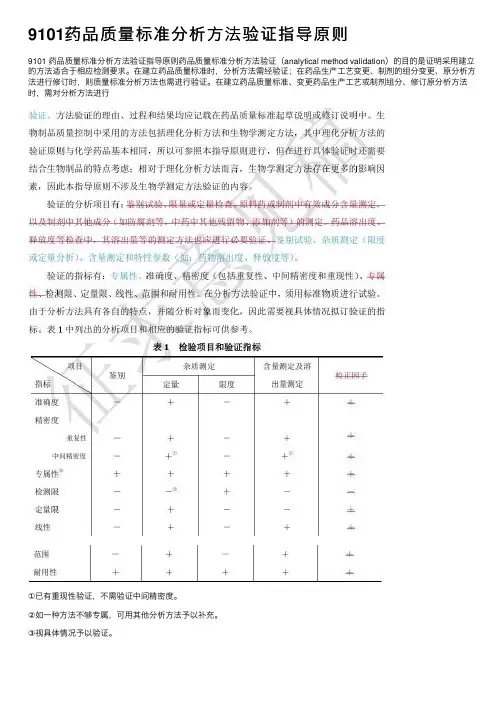
9101药品质量标准分析⽅法验证指导原则9101 药品质量标准分析⽅法验证指导原则药品质量标准分析⽅法验证(analytical method validation)的⽬的是证明采⽤建⽴的⽅法适合于相应检测要求。
在建⽴药品质量标准时,分析⽅法需经验证;在药品⽣产⼯艺变更、制剂的组分变更、原分析⽅法进⾏修订时,则质量标准分析⽅法也需进⾏验证。
在建⽴药品质量标准、变更药品⽣产⼯艺或制剂组分、修订原分析⽅法时,需对分析⽅法进⾏①已有重现性验证,不需验证中间精密度。
②如⼀种⽅法不够专属,可⽤其他分析⽅法予以补充。
③视具体情况予以验证。
⼀⼆、准确度准确度系指采⽤该所建⽴⽅法测定的结果与真实值或参⽐值接近的程度,⼀般⽤回收率(%)表⽰。
准确度应在规定的线性范围内测定试验。
准确度也可由所测定的精密度、线性和专属性推算出来。
1.化学药含量测定⽅法的准确度原料药采⽤对照品可⽤已知纯度的对照品或供试品进⾏测定,或⽤本法所得所测定结果与已知准确度的另⼀个⽅法测定的结果进⾏⽐较。
制剂可在处⽅量空⽩辅料中,加⼊已知量被测物对照品进⾏测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加⼊已知量的被测物进⾏测定,或⽤所建⽴⽅法的测定结果与已知准确度的另⼀种个⽅法测定结果进⾏⽐较。
准确度也可由所测定的精密度、线性和专属性推算出来。
对⾊谱⽅法⽽⾔,绝对(或定量)校正因⼦是指单位⾯积的⾊谱峰代表的待测物质的量。
待测定物质与所选定的参照物质的绝对校正因⼦之⽐,即为相对校正因⼦。
相对校正因⼦计算法常应⽤于化学药有关物质的测定、中药材及其复⽅制剂中多指标成分的测定。
校正因⼦的表⽰⽅法很多,本指导原则中的校正因⼦是指⽓相⾊谱法和髙效液相⾊谱法中的相对重量校正因⼦。
相对校正因⼦可采⽤替代物(对照品)和被替代物(待测物)标准曲线斜率⽐值进⾏⽐较获得;采⽤紫外吸收检测器时,可将替代物(对照品)和被替代物(待测物)在规定波长和溶剂条件下的吸收系数⽐值进⾏⽐较,计算获得。

一、目的:规范药品检验方法确认与验证的管理,证明采用的方法适合相应的检测要求及在实验室条件下的适用性,保证检验结果准确、可靠。
二、适用范围:适用于中药和化学药品(包括物料和产品)的理化分析方法和仪器分析方法的确认与验证。
不适用于中药和化学药品(包括物料和产品)微生物分析方法的确认与验证。
三、相关职责:QC班组长:QC班组长根据检验方法的来源确定开展方法确认或验证。
由其本人或者其他有经验的检验人员起草方案;负责确认或验证方案的培训;安排有经验的人员参与方法确认或验证过程实施;对确认或验证过程中出现的偏差要严格按照相关管理规程的程序执行,如需要提出变更申请;对确认或验证工作中出现的问题及时纠正并记录;总结确认或验证报告。
QC主管:负责审核检验方法确认或验证方案,并对方案的执行过程进行追踪;负责组织偏差的调查,变更的审核;负责总结报告的审核。
化验室QA:监督各项目按照已制定的方案进行;参与确认或验证过程中的偏差调查;对提出的变更进行评估,确认变更的是否成立,跟踪变更实施。
QA主管:负责审核检验方法确认或验证方案,确保其法规符合性;参与偏差的调查,变更的审核;负责总结报告的审核。
验证专员:审核检验方法确认或验证方案;审核总结报告;负责验证证书的发放;负责方法确认或验证方案、记录和报告的整理、存档。
质量管理负责人:批准检验方法确认或验证方案;批准方案实施过程中出现的偏差和变更;批准总结报告。
四、制定依据:《药品GMP指南》(质量控制实验室与物料系统)、《药品生产质量管理规范》(2010年修订)、《中国药典》(2020年版)、《化学药物质量控制分析方法验证技术指导原则》、ICH分析方法验证:正文和方法学Q2(R1 )、中国药品检验标准操作规范(2010年版)(原子吸收分光光度法)。
五、内容:1、术语1.1方法验证方法验证系指根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否符合检验项目的要求。

生物制品质量控制分析方法验证技术审评一般原则药品审评中心二〇〇五年十二月目录一、概述 (2)二、生物学测定常用方法 (3)三、方法的来源(种类) (4)四、分析方法 (5)五、分析方法的验证 (7)(一)专属性(二)准确性(三)精密度(四)线性(五)范围(六)耐用性(七)检测限度(八)定量限度六、综合分析 (13)七、名词解释 (13)八、参考文献 (14)九、附录(推荐的验证方案) (15)十、起草说明 (16)十一、著者 (21)一、概述质控分析方法验证就是证明采用的方法适合于相应检测要求,具有相当的准确性和可靠性,进而可以达到控制产品质量的目的。
只有经过验证的分析方法才能用于控制产品质量,因此方法验证是制定质量标准的基础。
一般情况下,需验证的分析项目有:鉴别试验、杂质检查、原液或制剂中有效成分的含量测定及生物活性测定。
其它质控方法,如必要时也应加以验证。
生物制品质控中采用的方法包括理化分析方法和生物学测定方法。
生物制品的理化分析方法验证原则与化学药品基本相同,可参照《化学药物质量控制分析方法验证技术指导原则》进行,同时需结合生物制品的特点考虑。
本技术审评一般原则主要针对生物学测定方法的验证进行讨论。
生物学测定方法可广泛用于各种检测目的,包括鉴别、生物活性和杂质检测等,其中最主要的是生物活性(或效价、效力)测定。
相对于理化分析方法而言,生物学测定具有更大的可变性,一般要使用动物、细胞或生物分子,因此对于生物学测定的判断标准可适当灵活掌握,但是对于定量测定方法应尽可能减少方法的变异,验证的结果仍应能证明该方法具有相当的准确性和可靠性,并应以能够有效控制产品质量为基本标准。
如未经必要的质控分析方法验证或由拟定质控分析方法构成的制造及检定规程难以控制产品质量,则对于所申报品种的质量可控性将无法评价。
本文在某些关键参数上提出一些原则性的建议或要求,拟作为技术审评的一般原则,同时也可供申报单位在进行相关研究工作时参考。
目的:方法验证的目的是证明采用的方法适合相应检测要求;方法确认目的是为确认方法在本实验室条件下的适用性。
应用范围:适用于本公司产品的检验方法。
编订依据:《中国药典》2010年版二部附录194页。
内容:1 分析方法的验证及确认的适用范围及发起时机2 方法验证方法验证就是根据检验项目的要求,预先设置一定的验证内容和验证标准要求,并通过设计合理的实验来验证所采用的分析方法是否答合检验项目的要求。
建立质量标准时,应对分析方法中的各检验项目进行完整的验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,可根据变更的内容决定对分析方法进行部分验证还是完整的验证;当原料药合成工艺发生变更时,可能引入新的杂质,杂质检查方法和含量测定方法的专属性就需要进行验证,以证明有关物质检查方法能够检测新引入的杂质,且新引入的杂质对主成分的含量测定应无干扰;当质量标准中某一项目分析方法发生部分改变时,如采用高效液相色谱法测定含量时,检测波长发生改变,则需要重新进行检测限、定量限、专属性、准确度、精密度、线内容的验证,证明修订后的分析方法的合理可靠;当变更达到一定程度时,则需要完整的验证。
如分析方法完全改奕,则应按新方法进行完整的验证。
2.1 方法验证的一般原则通常情况下,分析方法需进行方法验证。
对于仅需按照实验室日常测试操作步骤即可测定的检验项目不需要进行验证,如外观、崩解时限、密度、重量、pH值、灰分、装量等。
方法验证的内容应根据检验项目的要求,结合所采用分析方法的特点确定。
同一分析方法用于不同的检验项目会有不同的验证要求。
2.2 需要验证的检验项目检验项目是为控制药品的质量,保证药品安全有效而设定的测试项目。
根据检验项目的设定目的和验证内容的不同要求,一般需验证的检验项目分为四类:鉴别、杂质的限度检查、杂质的定量测定、含量测定包括原料药或制中有效成分的含量,制中其他成分的含量,溶出度与释放度等检查中的溶出量,以及含量均匀度。

药物制剂人体生物利用度和生物等效性试验指导原则人体生物利用度(bioavailability)是反映制剂中主药吸收进入人体体循环的相对量和速度的药代动力学参数。
生物等效性(bioequivalancy)指一种药物的不同制剂,在相同实验条件下以相同剂量用于人体,其吸收程度和速度无显著性差异。
两者概念不尽相同,但试验方法基本一致。
药物制剂人体生物利用度和生物等效性试验属临床试验范畴,故须具备我国药品临床试验管理规范要求的各项必要条件,并按规范要求进行试验。
一、生物样品分析方法的建立和验证应根据所试药物的理化特性建立生物样品的分析方法。
所建方法须经过充分验证,证明符合如下基本要求,方可用于正式试验。
(一)特异性须证明所测定物质是受试药品的原形药物或特定活性代谢物。
生物样品所含内源性物质或代谢物不得干扰对样品所测物质的分析。
根据药物结构特性,首选色谱法,如HPLC、GC、GC-MS或LC-MS等方法,并确定保证分析方法特异性的最佳条件。
色谱法应提供空白生物样品、标准品、空白生物样品加入标准品及用药后生物样品的色谱图,以反映分析方法的特异性。
(二)标准曲线与线性范围所测定物质的浓度与响应的相关性,用回归分析方法(如用加权最小二乘法)所得的回归方程来评价。
标准曲线高低浓度范围为线性范围,在线性范围内浓度测定结果应可达到试验要求的精密度和准确度。
必须用至少5个浓度建立标准曲线,应使用与待测样品相同生物介质,线性范围要能覆盖全部待测浓度,不得用线性范围外推的方法求算未知样品的浓度。
标准曲线不包括零点。
(三)精密度与准确度要求选择三个浓度的质控样品同时进行方法的精密度和准确度考察,低浓度选择在最低量限(LOQ)附近,高浓度在标准曲线的上限附近,中间选一个浓度,每一浓度至少测定5个样品。
精密度用质控样品的日内和日间相对标准差(RSD)表示,一般RSD应小于15%在LOQ附近RSD应小于20%。
准确度是指用特定方法测得的生物样品浓度与真实浓度的接近程度,可用相对回收率表示,一般应85%~115%范围内,在LOQ附近应在80%~120%范围内。

中药制剂溶出方法建立一、步骤1.选择适当的试剂:根据中药制剂的特点和药理活性成分的溶出特性,选择适当的试剂。
常见的试剂有水、生理盐水、人工胃肠液等。
2.确定试剂的pH值:试剂的pH值对药物的离解度和溶出速率有重要影响。
通过调节试剂的pH值,可以模拟不同环境中的药物溶出情况。
3.确定试剂的体积和搅拌速度:试剂的体积和搅拌速度对药物的溶出速率也有影响。
试剂的体积应尽量达到药物完全溶解的程度,搅拌速度应适当,以模拟人体胃肠道中的条件。
4.确定溶出仪器和方法:根据所需测试的溶出速度,选择合适的溶出仪器和方法。
常见的溶出仪器有柱式溶出仪、碟式溶出仪、流通系统等。
5.确定样品的质量和数量:样品的质量和数量对溶出方法的建立也具有重要意义。
样品的质量应符合法定标准,数量应足够进行多次重复实验。
6.进行实验:将样品放入溶出仪器中,根据已确定的方法进行试验。
在一定时间间隔内,取出溶出液进行分析,得到药物浓度-时间曲线。
7.建立溶出曲线:将所得的药物浓度-时间数据进行计算和统计,得到溶出曲线。
通常采用累加溶出率和瞬时溶出率来评价药物的溶出速度。
二、重要性1.评价中药制剂的质量:中药制剂的溶出性能与其药效密切相关。
通过建立溶出方法,可以评价中药制剂在体内的药物释放性能,进而评价其质量。
2.比较中药制剂的溶出性能:不同的中药制剂溶出性能可能存在差异,通过建立溶出方法,可以比较不同制剂剂型的药物释放速度,选择最适合的制剂。
3.研究中药制剂的释放机制:通过分析溶出曲线和溶出动力学参数,可以研究药物的释放机制,为制剂的改进和优化提供理论依据。
4.辅助制剂选择:一些中药制剂可能需要辅助溶出剂来促进药物的溶解和溶出,通过建立溶出方法,可以评价不同辅助溶出剂的效果,为制剂的配方设计提供参考。
三、案例分析以中药制剂为例,需要建立其溶出方法。
首先选择适当的试剂,根据该制剂的溶出特性和药理活性成分的特点,选择生理盐水作为试剂。
根据制剂的溶出特性需在不同pH条件下进行试验,确定试剂的pH值为1.2、4.5和6.8、然后确定试剂的体积为900 mL,搅拌速度为100 rpm。

体内药分作业J药剂0901 3091158012 钱晨药品质量标准分析方法验证的内容有:准确度,精密度(包括重复性,中间精密度和重现性),专属性,检测限,定量限,线性,范围和耐用性。
一、专属性1、体内:必须证明所测定的物质是原形药物或特定的活性代谢物,内源性物质和相应的代谢物不得干扰样品的测定。
对于色谱法至少要提供空白生物样品色谱图,空白生物样品外加对照色谱图记用药后的生物样品色谱图。
对于复方制剂应特别加强专属性研究,以排除可能的干扰。
2、体外:专属性系指在其他成分(如杂质,降解产物,辅料等)可能存在的情况下,采用的方法能正确测定出被测物的特性。
鉴别反应,杂质检查和含量测定方法,均应考察专属性。
如方法不够专属,应采用多个方法予以补充。
A、鉴别反应应能与可能共存的物质或结构相似的化合物区分。
不含被测物成分的供试品,以及结构相似或组分中的有关化合物,应均呈负反应。
B、含量测定和杂质测定色谱法和其他分离方法,应附代表性图谱,以说明方法的专属性,并标明诸成分在图谱中的位置,色谱中的分离应符合要求。
二、标准曲线和线性范围1、体内:根据所测定物质的浓度与响应的相关性,用回归分析方法获得标准曲线。
标准曲线高低浓度范围为线性范围,在线性范围内浓度测定结果应达到实验要求的精密度和准确度。
必须用至少6个浓度建立标准曲线,应使用与待测样品相同的生物介质,线性范围要能覆盖全部待测浓度,不允许将线性范围外推求算未知样品的浓度。
标准曲线不包括零点。
标准曲线上各浓度点的实测值与标示值的偏差(bias)在可接受范围内时,可判定标准曲线合格。
回归值系将各浓度点的响应值代人标准曲线计算所得的浓度值;标示值系指制备标准曲线时,各相应浓度点的配制浓度,标准曲线上各浓度点偏差的可接受范围一般规定为:最低浓度点的偏差在±20%以内,在其余各浓度点的偏差在±15%以内。
只有合格的标准曲线才能用于临床待测样品的浓度计算。
当线性范围较宽时,推荐采用加权最小二乘法(weighted least square method)进行同归计算2、体外:线性是只在设计范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。
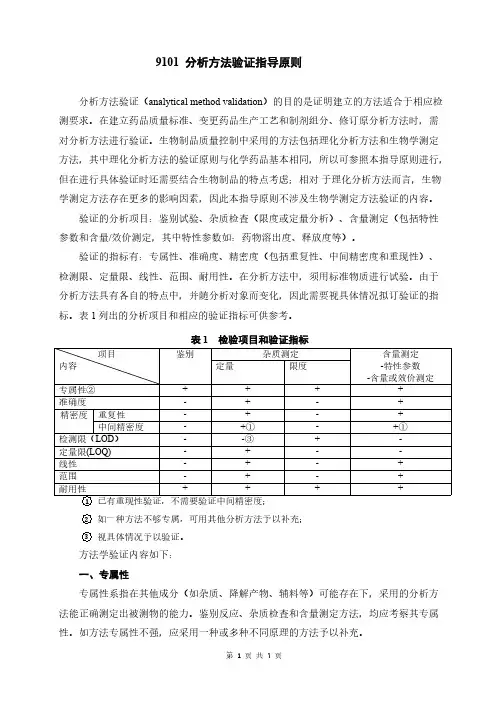
9101分析方法验证指导原则分析方法验证(analytical method validation)的目的是证明建立的方法适合于相应检测要求。
在建立药品质量标准、变更药品生产工艺和制剂组分、修订原分析方法时,需对分析方法进行验证。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目:鉴别试验、杂质检查(限度或定量分析)、含量测定(包括特性参数和含量/效价测定,其中特性参数如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、检测限、定量限、线性、范围、耐用性。
在分析方法中,须用标准物质进行试验。
由于分析方法具有各自的特点中,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表1列出的分析项目和相应的验证指标可供参考。
表1检验项目和验证指标项目内容鉴别杂质测定含量测定-特性参数-含量或效价测定定量限度专属性②++++准确度-+-+精密度重复性-+-+中间精密度-+①-+①检测限(LOD)--③+-定量限(LOQ)-+--线性-+-+范围-+-+耐用性++++ 1已有重现性验证,不需要验证中间精密度;2如一种方法不够专属,可用其他分析方法予以补充;3视具体情况予以验证。
方法学验证内容如下:一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
鉴别反应、杂质检査和含量测定方法,均应考察其专属性。
如方法专属性不强,应采用一种或多种不同原理的方法予以补充。
1.鉴别反应应能区分可能共存的物质或结构相似的化合物。
不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。



【S】GPH1-1生物制品质量控制分析方法验证技术审评一般原则药品审评中心2005年12月目 录一、概述 (2)二、生物学测定常用方法 (3)三、方法的来源(种类) (4)四、分析方法 (5)五、分析方法的验证 (7)(一)专属性(二)准确性(三)精密度(四)线性(五)范围(六)耐用性(七)检测限度(八)定量限度六、综合分析 (13)七、名词解释 (13)八、参考文献 (14)九、附录(推荐的验证方案) (15)十、起草说明 (16)十一、著者 (21)一、概述质控分析方法验证就是证明采用的方法适合于相应检测要求,具有相当的准确性和可靠性,进而可以达到控制产品质量的目的。
只有经过验证的分析方法才能用于控制产品质量,因此方法验证是制定质量标准的基础。
一般情况下,需验证的分析项目有:鉴别试验、杂质检查、原液或制剂中有效成分的含量测定及生物活性测定。
其它质控方法,如必要时也应加以验证。
生物制品质控中采用的方法包括理化分析方法和生物学测定方法。
生物制品的理化分析方法验证原则与化学药品基本相同,可参照《化学药物质量控制分析方法验证技术指导原则》进行,同时需结合生物制品的特点考虑。
本技术审评一般原则主要针对生物学测定方法的验证进行讨论。
生物学测定方法可广泛用于各种检测目的,包括鉴别、生物活性和杂质检测等,其中最主要的是生物活性(或效价、效力)测定。
相对于理化分析方法而言,生物学测定具有更大的可变性,一般要使用动物、细胞或生物分子,因此对于生物学测定的判断标准可适当灵活掌握,但是对于定量测定方法应尽可能减少方法的变异,验证的结果仍应能证明该方法具有相当的准确性和可靠性,并应以能够有效控制产品质量为基本标准。
如未经必要的质控分析方法验证或由拟定质控分析方法构成的制造及检定规程难以控制产品质量,则对于所申报品种的质量可控性将无法评价。
本文在某些关键参数上提出一些原则性的建议或要求,拟作为技术审评的一般原则,同时也可供申报单位在进行相关研究工作时参考。
2020版药典分析方法验证指导原则分析方法验证(analytical method validation)的目的是证明建立的方法适合于相应检测要求。
在建立药品质量标准、变更药品生产工艺或制剂组分、修订原分析方法时,需对分析方法进行验证。
生物制品质量控制中采用的方法包括理化分析方法和生物学测左方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑: 相对于理化分析方法而言,生物学测左方法存在更多的影响因素,因此本指导原则不涉及生物学测圧方法验证的内容。
验证的分析项目有:鉴别试验、杂质测立(限度或泄量分析)、含量测左(包括特性参数和含量/效价测立,苴中特性参数如:药物溶出度、释放度等)。
验证的指标有:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、龙量限、线性、范围和耐用性。
在分析方法验证中,须用标准物质进行试验。
由于分析方法具有各自的特点,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表1中列岀的分析项目和相应的验证指标可供参考。
表1检验项目和验证指标越表1检验项目和验证指标① 已有重现性验证,不需验证中间精密度。
②如一种方法不够专属,可用其他分析方法予以补充。
③ 视具体情况予以验证。
方法验证内容如下。
一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
鉴別反应、杂质检查和含量测泄方法,均应考察其专属性。
如方法专属性不强,应采用一种或多种不同原理的方法予以补充。
1.鉴别反应应能区分可能共存的物质或结构相似的化合物。
不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。
2.含量测泄和杂质测圮采用的色谱法和英他分离方法,应附代表性图谱,以说明方法的专属性,并应标明诸成分在图中的位置,色谱法中的分离度应符合要求。
任杂质对照品可获得的情况下,对于含量测怎,试样中可加入杂质或辅料,考察测左结果是否受干扰,并可与未加杂质或辅料的试样比较测圧结果。
复方丹参片检验方法验证方案一、背景然而,由于复方丹参片是一种复杂的中药制剂,其中含有多种不同成分。
因此,为保证复方丹参片的质量和疗效,需要建立一套有效的检验方法验证方案。
二、目的本方案的目的是建立适用于复方丹参片的常规质量控制方法,以验证复方丹参片的质量。
三、验证项目本方案中包含了以下几个验证项目:1.外观观察:包括复方丹参片的色泽、形状、大小等。
2.配方一致性验证:根据药材组分和比例,验证复方丹参片的配方是否符合规定。
3.质量标准验证:验证复方丹参片的含量测定方法是否标准、准确。
4.含量一致性验证:根据复方丹参片的主要有效成分,验证其含量在不同批次之间的一致性。
5.常规理化指标验证:包括总灰分、酸不溶性灰分、挥发性物质、水分含量、重金属等指标的检测。
四、验证方法1.外观观察:使用肉眼观察复方丹参片的色泽、形状、大小等,并与标准样品进行比较。
2.配方一致性验证:按照复方丹参片的配方,精确称取不同药材的重量,通过HPLC等方法分析验证各药材的含量是否符合配方要求。
3.质量标准验证:使用HPLC等分析法测定复方丹参片中主要有效成分的含量,与标准值进行对比。
4.含量一致性验证:选择多个不同批次的复方丹参片,使用HPLC等分析法测定其主要有效成分的含量,比较各批次之间的一致性。
5.常规理化指标验证:根据中药饮片行业标准,使用相应的检测方法对复方丹参片的总灰分、酸不溶性灰分、挥发性物质、水分含量、重金属等指标进行检测。
五、验证结果分析1.外观观察结果应与标准样品保持一致。
2.配方一致性验证结果应符合复方丹参片的配方要求。
3.质量标准验证结果应与标准值相近。
4.含量一致性验证结果应表明复方丹参片的主要有效成分在不同批次之间的含量一致性较好。
5.常规理化指标验证结果应符合中药饮片行业标准。
六、结论通过对复方丹参片的检验方法验证,可以得出以下结论:1.复方丹参片的外观符合标准要求。
2.复方丹参片的配方一致性良好,符合规定。