GCP对比学习-第八章必备文件管理
- 格式:pdf
- 大小:83.41 KB
- 文档页数:4

药物临床试验质量管理规范(修订草案征求意见稿)第一章总则第一条为保证药物临床试验过程规范,数据和结果的科学、真实、可靠,保护受试者的权益和安全,根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》,参照国际公认原则,制定本规范。
本规范适用于为申请药品注册而进行的药物临床试验。
药物临床试验的相关活动应当遵守本规范。
其他临床试验可参照本规范执行。
第二条药物临床试验质量管理规范(GCP)是药物临床试验全过程的标准规定,包括方案设计、组织实施、监查、稽查、记录、分析、总结和报告。
第三条药物临床试验应当符合《世界医学大会赫尔辛基宣言》原则及相关伦理要求,受试者的权益和安全是考虑的首要因素,优先于科学和社会获益。
伦理审查与知情同意是保障受试者权益的主要措施。
第四条药物临床试验应当有充分的科学依据。
临床试验应当权衡受试者和社会预期的风险和获益,只有当预期的获益大于风险时,方可实施或者继续临床试验。
第五条试验方案应当清晰、详细、可操作。
试验方案在获得伦理委员会同意后方可执行。
第六条研究者在临床试验过程中应当遵守试验方案,凡涉及医学判断或临床决策应当由临床医生做出。
参加临床试验实施的研究人员,应当具有在临床试验中承担相应工作的教育、培训和经验。
第七条所有临床试验的纸质或电子资料应当被妥善地记录、处理和保存,能够准确地报告、解释和确认。
应遵循临床试验中受试者隐私和保密性的法律法规,保护鉴别受试者身份的记录。
第八条试验药物的制备应当符合临床试验用药物生产质量管理相关规范。
试验药物的使用应当符合试验方案。
第九条临床试验的质量管理体系应当覆盖临床试验的全过程,以确保受试者保护、试验数据和结果的可靠性,以及临床试验遵守相关法律法规。
第十条临床试验的实施应当回避重大利益冲突。
第二章术语及其定义第十一条本规范下列用语的含义是:(一)临床试验(Clinical Trial),指以人体(病人或健康受试者)为对象的试验、研究,意在发现或验证某种试验药物的临床医学、药理学、其他药效学作用、不良反应,或者试验药物的吸收、分布、代谢和排泄,以确定药物的疗效与安全性的系统性试验。


GCP检查应知应会——资料管理员1. 资料文档管理注意哪些?答:临床试验资料管理贯穿整个临床试验过程中,临床试验资料必须做到准确、全面、规范及可溯源,研究者及时完整的如实记录、质控员认真全面的数据核查、文件管理人员周期性的归属是药物临床试验档案资料完整的重要保证。
因此,文件资料管理从试验启动会开始就应建立时时归属的概念,在试验启动阶段、执行阶段、总结阶段及时整理、查缺补漏。
1)交接:档案接收时,交接人与接收人要逐一清点核实档案,在档案交接记录表上双方签字;2)保密:存放临床试验文件柜应上锁,机构办公室及所有接触方案资料的人员均必须遵守国家保密法规,对申办者提供的方案、有关新处方、制剂工艺等关键内容保密,不得擅自对外泄露;3)借阅:阅览资料须经机构办主任同意,除涉及试验的研究者、申办者及试验质量监控人员等GCP规定查阅资料人员外,其他任何机构和个人不得擅自查阅试验相关文件,机构资料管理员负责文档存取,办理阅览手续;4)日常管理:整理、登记、分类、归档管理。
认真执行防护措施,符合防盗、防火、防虫、防鼠、防潮、防尘、防高温、防晒等要求;配备防潮和灭火装置,定期检查以确保正常运转;严禁放置易燃易爆品,不准吸烟;定期进行通风除湿,保证文件资料的安全。
2. 资料管理室防火情况,用什么灭火器?答:干粉灭火器,干粉灭火器灭火后会对档案资料有一定的污染作用,且难以清洁,后续可配备手提二氧化碳、手提七氟丙烷等洁净气体灭火器来辅助灭火。
3. 自动喷淋怎么办,档案柜金属可以防喷淋(现场查看)?答:无喷淋系统。
4. 临床试验相关材料的保存时间?答:用于申请药品注册的临床试验,必备文件应当至少保存至试验药物被批准上市后5年。
未用于申请药品注册的临床试验,必备文件应当至少保存至临床试验终止后5年。
或按照相关约定延长保存时间。
5. 资料保存有什么规定和要求?答:温度控制在14~24℃之间,相对湿度控制在45~60%之间。
符合防盗、防火、防虫、防鼠、防潮、防尘、防高温、防晒等要求。


gcp资料管理制度gcp资料管理制度篇一:药物临床试验文件管理制度药物临床试验文件管理制度1. 临床试验文件资料归档和保存应由科室药物临床试验文档管理员负责。
2. 文件包括:管理文件:包括机构及专业的管理制度、设计规范、标准操作规程、相关法律法规、研究人员资料和培训档案等。
临床试验文件是用于证明临床试验数据及临床操作的真实、准确、可靠的证据,包括临床试验前、试验中、试验后资料。
已完成的试验资料应及时交机构办公室归档,不得随意外借、随处摆放,以免丢失遗漏。
临床试验必须保存的文件资料的项目,可参照现行GCP附录部分所列出的必须保存的最少文件清单。
3. 临床试验文件资料档案:为便于管理和查阅,可以将每项临床试验的文件资料进行分类管理,主要类别如下:a)试验方案及修正案、批文; b)研究者手册及更新件;c)知情同意书及相关资料;d)病例报告表(样表及已填写的病例报告表);e)标准操作规程及更新版本;f)标准操作规程培训及分发、领用记录;g)与药品监督管理部门的沟通文件;h)与伦理委员会的沟通文件;i)与申办者、监督员的沟通文件;j)试验用药物管理文件; k)受试者招募、筛选及入选资料; l)不良事件记录及报告文件;m)研究人员名单及履历表;n)临床试验原始资料;o)其他临床试验相关文件资料。
4. 临床试验文件资料保存:保存期限:研究者在药物临床试验结束后2月内,应将资料归档交给医院机构办公室保存,研究者应保存临床试验资料至临床试验结束后至少5年;申办者应保存临床试验资料至试验药品批准上市后至少5年。
保存条件:临床试验文件资料应保存在专用且安全的场所,如专门的档案室;保存场所的温湿度应符合要求,而且具有防潮、防火、防丢失的设施,能保证文件资料的安全。
保存形式:可以纸质文件、电子记录、移动硬盘、刻录CD等形式保存。
5. 文件资料的保存必须建立完善的登记记录。
文件资料的查阅仅限临床试验的主要研究者、官方检查人员和相关试验项目申办者委派的稽查员。

各位临床研究的朋友大家下午好,这次我继续给大家讲新版ICH-GCP第八部分临床试验必需文件是能够体现出临床研究质量,对临床研究进行评估,同时体现及时的对临床研究文件进行整理,可以极大的帮助研究者、申办方和监查员很好ICH-GCP第八部分列出了所有的临床试验必需文件清单,根据试验的不同阶段总文件夹(Trial master files)在临床试验开始之前就应该建立起来。
临床试验ADDENDUM部分是新版ICH-GCP新增加的,体现在以下几个方面:申办者应当确保研究者可以控制并能继续访问CRF里的信息,申办方不能把在研究计划阶段,应产生下列文件并在试验正式开始之前归档。
下图(a)记录图(a)下图(b)记录着8.2.4,8.2.5和8.2.6的文件清单和对应的存档地点。
图(b)下图(c)记录着8.2.7的文件清单和对应的存档地点。
图(c)下图(d)记录着8.2.8,8.2.9,8.2.10和8.2.11的文件清单和对应的存档地点。
下图(e)记录着8.2.12,8.2.13,8.2.14和8.2.15的文件清单和对应的存档地图(e)下图(f)记录着8.2.16,8.2.17的文件清单和对应的存档地点。
图(f)下图(g)记录着8.2.18,8.2.19和8.2.20的文件清单和对应的存档地点。
图(g)临床试验进行期间,下列文件也应添加在档案中。
下图(h)记录着8.3.1的文件清单和对应的存档地点。
图(h)下图(i)记录着8.3.2和8.3.3的文件清单和对应的存档地点。
图(i)P21图(j)下图(k)记录着8.3.9,8.3.10,8.3.11,8.3.12和8.3.13的文件清单和对应的图(k)下图(l)记录着8.3.14,8.3.15,8.3.16,8.3.17,8.3.18和8.3.19的文件清单图(l)下图(m)记录着8.3.20,8.3.21,8.3.22,8.3.23,8.3.24和8.3.25的文件清图(m)在试验完成或中止之后,8.2及8.3节所列文件及下列文件皆应归档。
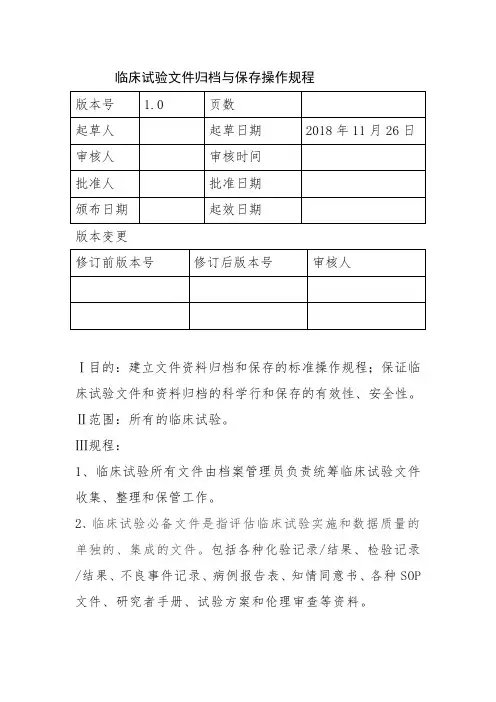
临床试验文件归档与保存操作规程版本变更Ⅰ目的:建立文件资料归档和保存的标准操作规程;保证临床试验文件和资料归档的科学行和保存的有效性、安全性。
Ⅱ范围:所有的临床试验。
Ⅲ规程:1、临床试验所有文件由档案管理员负责统筹临床试验文件收集、整理和保管工作。
2、临床试验必备文件是指评估临床试验实施和数据质量的单独的、集成的文件。
包括各种化验记录/结果、检验记录/结果、不良事件记录、病例报告表、知情同意书、各种SOP 文件、研究者手册、试验方案和伦理审查等资料。
3、临床试验文件资料保存:临床试验的准备、进行和结束过程中,相关文件存放于专用文件储存柜;保存场所的温湿度应符合要求,而且具有防潮、防火、防丢的设施,能保证文件资料的安全。
4、文件保存形式:用于保存临床试验资料的介质应当确保源数据或其真实副本在留存期内保存完整和可读取,并定期测试或检查恢复读取的能力,免于被故意或无意地更改或丢失。
可以纸质文件、电子记录、移动硬盘、刻录CD等形式保存。
5、文件保存时间:用于申请药品注册的临床试验,必备文件应当至少保存至试验药物被批准上市后2年;未用于申请药品注册的临床试验,必备文件应当至少保存至临床试验终止后5年。
6、文件资料的保存必须建立完善的登记记录。
登记情况见附录1。
档案管理员及相关研究人员须遵守国家保密法规,除涉及临床试验的研究者、申办方、第三方稽查与官方视察人员外,其他任何机构和个人不得擅自查阅试验相关文件,所有研究人员查阅、取用相关文件时必须登记原因及时间并签字,使用完后立即归还。
与研究无关人员不得查阅、取用试验文件。
参考文献:国家食品药品监督管理局令第3号发布《药品临床试验管理规范》,2003,第八章附录1临床试验项目文件归档资料临床试验资料归档清单备注:相应材料暂未递交时,勾选“无”;如该项不适用,例如不需要招募广告的项目,则相应内容勾选“NA”。
如未归档,在备注栏填上原因。



第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
第八章文件管理
品质管理
品质管理第八章文件管理
品质管理
品质管理第八章文件管理
品质管理第八章文件管理
品质管理
第八章文件管理
品质管理
品质管理第八章文件管理
品质管理第八章文件管理
第八章文件管理
品质管理
品质管理。
GCP对比学习-第八章必备文件管理第八章必备文件管理-管理要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第七十七条临床试验必备文件管理的要求临床试验必备文件是指评估临床试验实施和产生数据的单独的、集成的、质量可控的文件。
这些文件用于证明研究者、申办者和监查员在临床试验过程中,遵守了本规范和相关药物临床试验的法律法规要求。
必备文件是申办者独立稽查、药品监督管理部门检查临床试验的主要内容,并作为确认临床试验实施的真实性和所收集数据完整性的依据。
第七十九条临床试验必备文件是指评估临床试验实施和数据质量的单独的、集成的文件。
这些文件用于证明研究者、申办者和监查员在临床试验过程中遵守了本规范和相关药物临床试验的法律法规要求。
必备文件是申办者稽查、药品监督管理部门检查临床试验的重要内容,并作为确认临床试验实施的真实性和所收集数据完整性的依据。
8. ESSENTIAL DOCUMENTS FOR THECONDUCT OF A CLINICAL TRIAL8.1 Introduction Essential Documents are thosedocuments which individually and collectivelypermit evaluation of the conduct of a trial and thequality of the data produced. These documents serveto demonstrate the compliance of the investigator,sponsor and monitor with the standards of GoodClinical Practice and with all applicable regulatoryrequirements. Essential Documents also serve anumber of other important purposes. Filing essentialdocuments at the investigator/institution and sponsorsites in a timely manner can greatly assist in thesuccessful management of a trial by the investigator,sponsor and monitor. These documents are also theones which are usually audited by the sponsor'sindependent audit function and inspected by theregulatory authority(ies) as part of the process toconfirm the validity of the trial conduct and theintegrity of data collected.学习笔记sunhua-yjsyy整理第八章必备文件管理-保存要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第七十八条必备文件保存的要求申办者、研究者及其供职的医疗机构应确认,双方均有保存这些临床试验必备文件的场所和条件。
【对比】-新版GCP必备文件和ICH的有啥不一样?
在新版GCP发布之后,大家就一直有个疑惑,GCP中明明提到了必备文件,但是为什么没有对应的附录呢?现在终于来了。
附件中明确提到参考的文献是:
ICH E6(R2) Guideline for good clinical practice
那么让我们一起来看看新版GCP必备文件和ICH的有啥不一样吧!
先说结论:
1.框架及条目基本一致;
2.部分在ICH中为“必要时”的文件,在新版GCP中为必须:伦理委员会成员名单(申办方需要存)、监管部门的批件和备案(申办方和机构都需要存);
3.研究者简历收集新版GCP要求收集所有授权的人员签字版简历及资质。
GCP中提到必备文件的部分条款:
第十一条本规范下列用语的含义是:
(三十三)必备文件,指能够单独或者汇集后用于评价临床试验的实施过程和试验数据质量的文件。
第二十五条试验的记录和报告应当符合以下要求:
(四)研究者和临床试验机构应当按“临床试验必备文件”和药品监督管理部门的相关要求,妥善保存试验文档。
第三十六条申办者在试验管理、数据处理与记录保存中应当符合以下要求
(八)申办者应当保存与申办者相关的临床试验数据,有些参加临床试验的相关单位获得的其他数据,也应当作为申办者的特定数据保留在临床试验必备文件内
第五十条监查员的职责包括:
(十)监查员确认研究者是否按照本规范保存了必备文件。
第八章必备文件管理
下面是ICH与2020版GCP的详细对照:。
第1篇一、总则为规范公司Google Cloud Platform(GCP)相关资料的管理,确保资料的安全、完整和有效利用,提高工作效率,特制定本制度。
二、适用范围本制度适用于公司内部所有涉及GCP的资料,包括但不限于技术文档、项目方案、用户手册、操作指南、数据报表、配置文件等。
三、资料分类1. 公开资料:对内公开的资料,如产品介绍、技术白皮书等。
2. 内部资料:仅限于公司内部使用的资料,如项目方案、内部培训资料等。
3. 机密资料:涉及公司商业秘密、客户信息、技术核心等内容的资料。
四、资料管理职责1. 资料管理部门:负责制定资料管理制度,监督执行,定期评估资料管理效果。
2. 资料责任人:负责所负责资料的分类、收集、整理、归档和保密工作。
3. 使用人员:负责按照规定使用资料,不得泄露、篡改或非法复制。
五、资料收集与整理1. 资料收集:资料责任人应按照资料分类要求,及时收集各类GCP资料。
2. 资料整理:资料责任人应对收集到的资料进行分类、整理、归档,确保资料完整、清晰、易于检索。
六、资料存储与备份1. 存储介质:资料应存储在安全可靠的介质上,如电子文档、纸质文档等。
2. 备份:定期对资料进行备份,确保资料不因硬件故障、人为失误等原因丢失。
七、资料保密1. 保密要求:机密资料应严格按照保密规定进行管理,未经授权不得泄露。
2. 访问控制:对机密资料设置访问权限,仅授权人员可查阅和使用。
3. 安全措施:采取物理、技术、管理等措施,确保资料安全。
八、资料使用与借阅1. 使用规范:使用人员应按照资料内容和使用目的进行查阅和使用,不得擅自复制、传播或篡改。
2. 借阅:需借阅资料的人员应填写借阅申请,经资料责任人批准后方可借阅。
九、资料更新与维护1. 更新:资料责任人应定期对资料进行更新,确保资料内容的准确性和时效性。
2. 维护:定期对资料进行维护,确保资料存储介质的完好和资料检索系统的正常运行。
十、监督检查1. 自查:资料管理部门应定期组织自查,检查资料管理制度执行情况。
第八章必备文件管理-管理要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第七十七条临床试验必备文件管理的要求临床试验必备文件是指评估临床试验实施和产生数据的单独的、集成的、质量可控的文件。
这些文件用于证明研究者、申办者和监查员在临床试验过程中,遵守了本规范和相关药物临床试验的法律法规要求。
必备文件是申办者独立稽查、药品监督管理部门检查临床试验的主要内容,并作为确认临床试验实施的真实性和所收集数据完整性的依据。
第七十九条临床试验必备文件是指评估临床试验实施和数据质量的单独的、集成的文件。
这些文件用于证明研究者、申办者和监查员在临床试验过程中遵守了本规范和相关药物临床试验的法律法规要求。
必备文件是申办者稽查、药品监督管理部门检查临床试验的重要内容,并作为确认临床试验实施的真实性和所收集数据完整性的依据。
8. ESSENTIAL DOCUMENTS FOR THECONDUCT OF A CLINICAL TRIAL8.1 Introduction Essential Documents are thosedocuments which individually and collectivelypermit evaluation of the conduct of a trial and thequality of the data produced. These documents serveto demonstrate the compliance of the investigator,sponsor and monitor with the standards of GoodClinical Practice and with all applicable regulatoryrequirements. Essential Documents also serve anumber of other important purposes. Filing essentialdocuments at the investigator/institution and sponsorsites in a timely manner can greatly assist in thesuccessful management of a trial by the investigator,sponsor and monitor. These documents are also theones which are usually audited by the sponsor'sindependent audit function and inspected by theregulatory authority(ies) as part of the process toconfirm the validity of the trial conduct and theintegrity of data collected.学习笔记sunhua-yjsyy整理第八章必备文件管理-保存要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第七十八条必备文件保存的要求申办者、研究者及其供职的医疗机构应确认,双方均有保存这些临床试验必备文件的场所和条件。
保存文件的设备条件应具备防止光线直接照射、防水、环境利于长期文件的保存,并制定文件管理的规章制度和SOP。
被保存的文件需要易于识别、查找、调阅和归位。
临床试验过程中产生的一些文件,如果未列在临床试验各阶段建立的必备文件管理目录中,申办者、研究者及研究机构也需将其列入各自的必备文件档案中保存。
第八十条申办者、研究者和临床试验机构应当确认均有保存临床试验必备文件的场所和条件。
保存文件的设备条件应当具备防止光线直接照射、防水、防火等环境,有利于文件的长期保存。
应当制定文件管理的标准操作规程。
被保存的文件需要易于识别、查找、调阅和归位。
用于保存临床试验资料的介质应当确保源数据或其真实副本在留存期内保存完整和可读取,并定期测试或检查恢复读取的能力,免于被故意或无意地更改或丢失。
临床试验实施中产生的一些文件,如果未列在临床试验必备文件管理目录中,申办者、研究者及临床试验机构也可以根据必要性和关联性将其列入各自的必备文件档案中保存。
8.1 The sponsor and investigator/institution shouldmaintain a record of the location(s) of theirrespective essential documents including sourcedocuments. The storage system used during the trialand for archiving (irrespective of the type of mediaused) should provide for document identification,version history, search, and retrieval. Essentialdocuments for the trial should be supplemented ormay be reduced where justified (in advance of trialinitiation) based on the importance and relevance ofthe specific documents to the trial.第八章必备文件管理-保存时限要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第八十一条用于申请药品注册的临床试验,必备文件应当至少保存至试验药物被批准上市后2年;未用于申请药品注册的临床试验,必备文件应当至少保存至临床试验终止后5年。
新增,E6(R2)8.1项下未涉及学习笔记sunhua-yjsyy整理第八章必备文件管理-源数据、CRF数据的管理GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第七十九条必备文件中源数据、CRF数据的管理申办者应确保研究者能保留已递交给申办者的CRF数据。
用作源文件的复印件必须满足核证副本的要求。
研究者及其供职的医疗机构能够管理所有临床试验的必备文件,并且保证在临床试验实施全过程中原始数据产生的真实性。
第八十二条申办者应当确保研究者始终可以控制和查阅报告给申办者的病例报告表中的数据,该数据不应该只由申办者控制。
申办者应当确保研究者能保留已递交给申办者的病例报告表数据。
用作源文件的复印件应当满足核证副本的要求。
8.1 The sponsor should ensure that the investigatorhas control of and continuous access to the CRF datareported to the sponsor. The sponsor should not haveexclusive control of those data. When a copy is usedto replace an original document (e.g., sourcedocuments, CRF), the copy should fulfill therequirements for certified copies. Theinvestigator/institution should have control of allessential documents and records generated by theinvestigator/institution before, during, and after thetrial.第八章必备文件管理-分阶段保存的要求GCP-2016版征求意见稿GCP-2018版草案征求意见稿备注第八十条必备文件分阶段保存的要求必备文件根据临床试验不同阶段归类为三个阶段:临床试验准备阶段、临床试验进行阶段、临床试验完成后阶段。
(临床试验保存文件见附件)。
每一必备文件需说明其存在的目的,并说明该文件需要列入研究者及其供职的医疗机构保管,或申办者、或研究者和申办者双方保管的必第八十三条每个必备文件需要说明其存在的目的,并说明该文件需要列入研究者及临床试验机构保管、申办者保管或者双方保管的必备文件档案中。
临床试验开始时,研究者及临床试验机构、申办者双方均应当建立必备文件的档案管理。
临床试验结束时,监查员应当审核确认研究者及临床试验机构、申办者的必备文件,这些文件应当The minimum list of essential documents which hasbeen developed follows. The various documents aregrouped in three sections according to the stage ofthe trial during which they will normally begenerated: 1) before the clinical phase of the trialcommences, 2) during the clinical conduct of thetrial, and 3) after completion or termination of thetrial. A description is given of the purpose of each学习笔记sunhua-yjsyy整理备文件档案中。
临床试验开始时,研究者及其供职的医疗机构、申办者双方各自的办公室均应建立试验必备文件的档案管理。
试验结束时,监查员必须审核确认研究者及研究机构、申办者各自的必备文件,这些文件必须被妥善的保存在各自的临床试验档案卷宗内。
被妥善地保存在各自的临床试验档案卷宗内。
document, and whether it should be filed in either theinvestigator/institution or sponsor files, or both. It isacceptable to combine some of the documents,provided the individual elements are readilyidentifiable. Trial master files should be establishedat the beginning of the trial, both at theinvestigator/institution’s site and at the sponsor'soffice. A final close-out of a trial can only be donewhen the monitor has reviewed bothinvestigator/institution and sponsor files andconfirmed that all necessary documents are in theappropriate files. Any or all of the documentsaddressed in this guideline may be subject to, andshould be available for, audit by the sponsor’s auditorand inspection by the regulatory authority(ies)学习笔记sunhua-yjsyy整理。