第五章 高聚物的物理状态与特征温度
- 格式:pdf
- 大小:283.89 KB
- 文档页数:32



《塑料成型加工基础》单元电子教材高分子的特征温度一、高分子的特征温度高分子的特征温度是高聚物在外力作用时,随温度的变化引起的形变大小或性能发生转变时的温度。
高分子的特征温度主要有玻璃化温度(T g)、熔点(T m)、黏流温度(T f)、热分解温度(T d)、脆化温度(T b)、软化温度(T s)等。
二、玻璃化温度玻璃化温度是高聚物链段运动开始发生(或被冻结)的温度,用T g表示。
它是非晶高聚物作为塑料使用时的耐热温度(或最高使用温度)和作为橡胶使用的耐寒温度(或最低使用温度)。
三、影响玻璃化温度的因素①分子主链柔性的影响凡是对大分子主链柔性有影响影响因素,对玻璃化温度都有影响。
柔性越大,玻璃化温度越小。
②分子间作用力的影响分子间作用力越大,则玻璃化温度越高。
能够在分子间形成氢键的聚酰胺、聚乙烯醇、聚丙烯酸、聚丙烯腈等的玻璃化温度都较高。
③分子量的影响分子量对玻璃化温度的影响,可以参看图3-2曲线及相关的解释。
也可以用数学经验公式来表示:MK T T g g -=∞ 式中,T g ——高聚物的玻璃化温度; ∞g T ——分子量无限大时的玻璃化温度,实际上为与分子量有关的玻璃化温度上限值;K ——常数;M ——高聚物的平均分子量。
该式说明,玻璃化温度随高聚物平均分子量的增加而增大,当高聚物平均分子量的增加到一定数值后,玻璃化温度变化不大,并趋于某一定值。
④ 共聚的影响共聚物的玻璃化温度总是介于组成该共聚物的两个或若干个不同单体的均聚物玻璃化温度之间。
对于双组分无规共聚物的玻璃化温度通常可用下式表示:gB B gA A g T V T V T +=gBB gA A g T W T W T +=1 式中,T g —— 共聚物的玻璃化温度;T gA —— A 单体均聚物的玻璃化温度;T gB —— B 单体均聚物的玻璃化温度;V A 、V B —— A 、B 单体共聚时的体积分数;W A 、W B —— A 、B 单位共聚时的质量分数。
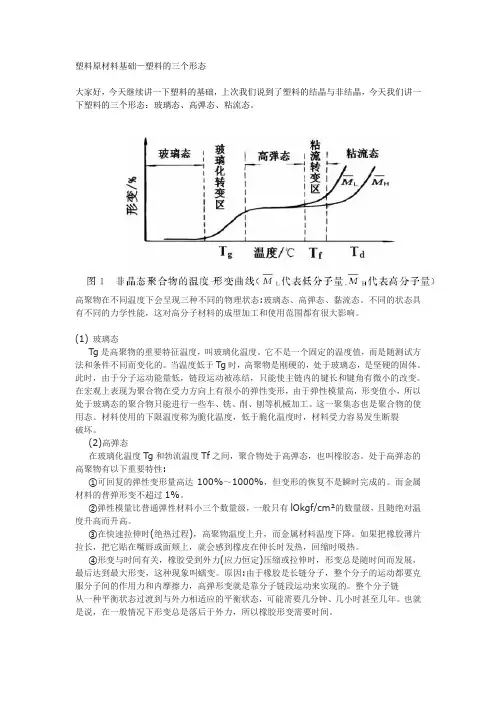
塑料原材料基础—塑料的三个形态大家好,今天继续讲一下塑料的基础,上次我们说到了塑料的结晶与非结晶,今天我们讲一下塑料的三个形态:玻璃态、高弹态、粘流态。
高聚物在不同温度下会呈现三种不同的物理状态:玻璃态、高弹态、黏流态。
不同的状态具有不同的力学性能,这对高分子材料的成型加工和使用范围都有很大影响。
(1)玻璃态Tg是高聚物的重要特征温度,叫玻璃化温度。
它不是一个固定的温度值,而是随测试方法和条件不同而变化的。
当温度低于Tg时,高聚物是刚硬的,处于玻璃态,是坚硬的固体。
此时,由于分子运动能量低,链段运动被冻结,只能使主链内的键长和键角有微小的改变。
在宏观上表现为聚合物在受力方向上有很小的弹性变形,由于弹性模量高,形变值小,所以处于玻璃态的聚合物只能进行一些车、铣、削、刨等机械加工。
这一聚集态也是聚合物的使用态。
材料使用的下限温度称为脆化温度,低于脆化温度时,材料受力容易发生断裂破坏。
(2)高弹态在玻璃化温度Tg和勃流温度Tf之间,聚合物处于高弹态,也叫橡胶态。
处于高弹态的高聚物有以下重要特性:①可回复的弹性变形量高达100%~1000%,但变形的恢复不是瞬时完成的。
而金属材料的普弹形变不超过1%。
②弹性模量比普通弹性材料小三个数量级,一般只有lOkgf/cm²的数量级,且随绝对温度升高而升高。
③在快速拉伸时(绝热过程),高聚物温度上升,而金属材料温度下降。
如果把橡胶薄片拉长,把它贴在嘴唇或面颊上,就会感到橡皮在伸长时发热,回缩时吸热。
④形变与时间有关,橡胶受到外力(应力恒定)压缩或拉伸时,形变总是随时间而发展,最后达到最大形变,这种现象叫蠕变。
原因:由于橡胶是长链分子,整个分子的运动都要克服分子间的作用力和内摩擦力,高弹形变就是靠分子链段运动来实现的。
整个分子链从一种平衡状态过渡到与外力相适应的平衡状态,可能需要几分钟、几小时甚至几年。
也就是说,在一般情况下形变总是落后于外力,所以橡胶形变需要时间。

聚合物的物理状态及其特点
聚合物是由大量重复单元组成的高分子化合物,广泛存在于自然界和人工合成中。
根据其物理状态不同,聚合物可以分为三种状态:固态、液态和气态。
每种状态都有其独特的特点和性质。
固态
在固态状态下,聚合物分子紧密排列,形成有序的晶格结构。
这种状态下的聚合物通常具有较高的密度和硬度,同时具有一定的弹性。
固态聚合物常见的形式包括晶体和非晶体两种。
晶体
固态聚合物在形成晶体结构时,分子呈有序排列,形成规则的晶格。
这种结构使得晶体聚合物具有较高的结晶度和机械性能,例如强度和硬度较高。
晶体聚合物还表现出明显的各向异性,即在不同方向上性能不同。
非晶体
非晶体聚合物则是指分子排列较为无序,没有明显晶格结构的聚合物状态。
这种状态下的聚合物通常具有较低的结晶度和强度,但具有较好的韧性和韧度。
非晶体聚合物在加工和改性方面具有一定的优势,因为其结构较为灵活。
液态
在液态状态下,聚合物分子之间相互滑动,没有固定的空间结构。
这种状态下的聚合物表现出类似于流体的性质,具有较高的流动性和可变形性。
液态聚合物常用于溶液、涂料、胶黏剂等领域。
气态
在气态状态下,聚合物的分子高度分散,并以气体形式存在。
气态聚合物通常具有较低的密度和较高的可压缩性,常见于气体分离、气体存储等领域。
总的来说,聚合物的物理状态在很大程度上影响着其性能和用途。
不同状态下的聚合物具有各自独特的特点,科学家们正通过不断的研究和改进,探索聚合物在不同状态下的性能和应用,为工程和科学领域带来更多可能性。
![[自然科学]第五章 高聚物的物理状态与特征温度](https://uimg.taocdn.com/6983be8fad02de80d5d840ad.webp)

以下为1~6章的名词解释,资料来源为高分子物理(第四版)材料科学基础(国外引进教材),化工大词典,百度百科,维基百科等。
第一章高分子链的结构全同立构:高分子链全部由一种旋光异构单元键接而成间同立构:高分子链由两种旋光异构单元交替键接而成构型:分子中由化学键所固定的原子在空间的几何排列,这种排列是热力学稳定的,要改变构型必需经过化学键的断裂与重组分子构造(Architecture):指聚合物分子的各种形状,一般高分子链的形状为线形,还有支化或交联结构的高分子链,支化高分子根据支链的长短可以分为短支链支化和长支链支化两种类型共聚物的序列结构:是指共聚物根据单体的连接方式不同所形成的结构,共聚物的序列结构分为四类:无规共聚物、嵌段共聚物、交替共聚物、接枝共聚物接枝共聚物:由两种或多种单体经接枝共聚而成的产物,兼有主链和支链的性能。
嵌段共聚物(block copolymer):又称镶嵌共聚物,是将两种或两种以上性质不同的聚合物链段连在一起制备而成的一种特殊聚合物。
环形聚合物:它的所有结构单元在物理性质和化学性质上都是等同的超支化聚合物:是在聚合物科学领域引起人们广泛兴趣的一种具有特殊大分子结构的聚合物构象:由于σ单键内旋转而产生的分子在空间的不同形态。
链段:高分子链上划分出的可以任意取向的最小单元或高分子链上能够独立运动的最小单元称为链段。
链柔性:是指高分子链在绕单键内旋转自由度,内旋转可导致高分子链构象的变化,因为伴随着状态熵增大,自发地趋向于蜷曲状态的特性。
近程相互作用:是指同一条链上的原子或基团之间,沿着链的方向,因为距离相近而产生相互作用远程相互作用:因柔性高分子链弯曲所导致的沿分子链远距离的原子或基团之间的空间相互作用。
远程相互作用可表现为斥力或引力,无论是斥力还是引力都使内旋转受阻,构想数减少,柔性下降,末端距变大。
自由连接链:假定分子是由足够多的不占体积的化学键自由结合而成,内旋转时没有键角限制和位垒障碍,其中每个键在任何方向取向的几率都相同。


第五章 聚合物的分子运动和转变1.聚合物分子运动的特点: ①.运动单元的多重性 ②.分子运动的时间依赖性 ③.分子运动的温度依赖性2.运动单元的多重性: A.具有多种运动模式 B.具有多种运动单元A.具有多种运动模式:由于高分子的长链结构,分子量不仅高,还具有多分散性,此外,它还可以带有不同的侧基,加上支化,交联,结晶,取向,共聚等,使得高分子的运动单元具有多重性,或者说高聚物的分子运动有多重模式B.具有多种运动单元:如侧基、支链、链节、链段、整个分子链等* 各种运动单元的运动方式①.链段的运动: 主链中碳-碳单键的内旋转, 使得高分子链有可能在整个分子不动,即分子链质量中心不变的情况下, 一部分链段相对于另一部分链段而运动②.链节的运动: 比链段还小的运动单元③.侧基的运动: 侧基运动是多种多样的, 如转动, 内旋转, 端基的运动等④.高分子的整体运动: 高分子作为整体呈现质量中心的移动⑤.晶区内的运动: 晶型转变,晶区缺陷的运动,晶区中的局部松弛模式等3.分子运动的时间依赖性: 在一定的温度和外力作用下, 高聚物分子从一种平衡态过渡到另一种平衡态需要一定时间的,这种现象即为分子运动的时间依赖性; 因为各种运动单元的运动都需克服内摩擦阻力, 不可能瞬时完成4.松弛现象:除去外力,橡皮开始回缩,其中的高分子链也由伸直状态逐渐过渡到卷曲状态,即松弛状态。
故该过程简称松弛过程。
5.松弛时间τ : 形变量恢复到原长度的1/e 时所需的时间 6.分子运动的温度依赖性:①.温度升高,使分子的内能增加:运动单元做某一模式的运动需要一定的能量, 当温度升高到运动单元的能量足以克服的能垒时,这一模式的运动被激发。
②.温度升高使聚合物的体积增加:分子运动需要一定的空间, 当温度升高到使自由空间达到某种运动模式所需要的尺寸后, 这一运动就可方便地进行。
7.黏弹行为的五个区域: ①.玻璃态 ②.玻璃化转变区 ③.高弹态(橡胶-弹性平台区) ④.粘弹转变区 ⑤.粘流态8.图- -:模量-温度曲线----各区的运动单元、特点、名字、描述玻璃化转变为高弹态,转变温度称为玻璃化温度Tg高弹态转变为粘流态,转变温度称为粘流温度Tf* 非晶聚合物:()()t -τΔx t =Δx 0e①.从相态角度来看,玻璃态,高弹态,粘流态均属液相,即分子间的相互排列均是无序的。

第五章高聚物的物理性能第一节高聚物的物理状态高聚物的聚集态结构,根据链结构的规整性和能否结晶可分为两类:.结晶性高聚物〔有规那么排列〕非结晶性高聚物〔无规那么排列〕"链段运动一一使高聚物具有高弹性高聚物热运动具有两重性<——个分子链运动一一使高聚物象液体一样具有粘流性热-机械曲线一一形变-温度曲线:表示高聚物材料在一定负荷下,形变大小与温度的关系曲线.按高聚物的结构可以分为:线型非晶高聚物形变-温度曲线结晶态高聚物形变-温度曲线其他类型的形变-温度曲线三种、线型非晶态高聚物的物理状态1 .形变-温度曲线Tb Tg 温度〔C〕TfTb—脆化温度;Tg —玻璃化温度;Tf—粘流温度可分为五个区A区〔玻璃态〕:内部结构类似玻璃,大分子不能运动,链段也不能运动,在除去外力后,形变马上消失而恢复原状, 可逆形变称为普弹性形变.C区〔高弹态或橡胶态〕:除了普弹形变外,主要发生了大分子的链段位移〔取向〕运动.但整个大分子间并未发生相对位移,形变也可以消除,所以是可逆的弹性形变.E区〔粘流态或塑化态〕:当施加负荷时,高聚物象粘性液体一样,发生分子粘性流动, 大分子能运动,链段也能运动,形变不能自动全部消除,这种不可逆特性,称为可塑性.B区和D区:为过渡区.其性质介于前后两种状态之间.C玻璃态物理力学三态j高弹态〔是一般非晶态高聚物所共有的〕粘流态2.状态力学行为特征形变机理应用玻璃态断裂伸长率<10%弹性模量大形变可逆〔普通形变〕键角、键长微改变链段微伸缩〔在原来平衡位置上发生位移〕塑料T b<T<T g高弹态断裂伸长率100~1000%弹性模量小形艾可逆链段发生位移橡胶T g<T<T f粘流态形变是/、可逆〔永久形变〕力学行为特征依赖大分子链段和大分子都发生位移黏合剂、油漆T f<T<T d注:.断裂伸长率断一原原2 .弹性模量:表示物体在拉压时,材料反抗弹性形变的水平.3 .三态之间的转变随温度的变化而逐渐变化过程玻璃态高弹态粘流态4 .注意问题1/ T g是大分子链段能运动的最低温度,高弹态的出现是链段运动的产物.2/ T g与柔性的关系:柔性大, T g低,反之.刚性大,T g高.3/ T g与T f的使用价值T g是塑料、纤维的最高使用温度T f是橡胶的最低使用温度,也是高聚物成型加工温度.5 .线型非晶态高聚物的物理力学状态与相对分子质量的关系不同相对分子质量的聚苯乙烯的热一机械曲线、结晶态高聚物的物理状态晶态高聚物的形变-温度曲线1 一一般相对分子质量2-相对分子质量很大1/结晶态高聚物按成型工艺条件的不同可以处于晶态和非晶态.2/晶态高聚物的形变-温度曲线可以分为①一般相对分子质量结晶高聚物只有两态:在T m以下处于晶态,这时与非晶态的的玻璃态相似,可以作塑料或纤维使用;在T m 以上时处于粘流态,可以进行成型加工.②相对分子质量很大高聚物有三种物理力学状态:温度在T m以下时为玻璃态,温度在T m与T f之间时为高弹态,温度在T f以上时为粘流态. 这时可以进行成型加工, 但由于高弹态一般不便成型加工, 而且温度高了又容易分解, 使成型产品的质量降低, 为此, 晶态高聚物的相对分子质量不宜太高.第二节各种特征温度与测定高聚物特征温度有:T g、T m、T f、T d、T b、T s一、玻璃化温度玻璃化转变:玻璃态向高弹态的转变, 是高聚物的链段运动被冻结或解冻而引起的聚集态转变〔不是热力学相变〕转变特征:发生时,高聚物的物理性能如比热、比容、导热系数、弹性模量、折光指数、介点常数、强度等都发生变化,故可用此来间接测T g.1 . 玻璃化温度的定义及应用定义:是tWj聚物链段运动开始发生〔或被冻结〕的温度, T g表示.应用:是非晶高聚物作为塑料使用时的耐热温度〔或最高使用温度〕和作为橡胶使用的耐寒温度〔或最低使用温度〕.2 .影响玻璃化温度的因素⑴大分子主链柔性的影响柔性T , T g J⑵分子间作用力的影响分子间作用力的影响T ,那么T gf.⑶相对分子质量的影响数学经验公式来表示:T g = T g" —K / M n说明:M小时M nf , T gfM n 较大时, T g 变化不大,并趋于某一定值.⑷共聚的影响共聚物的玻璃化温度总是介于组成该共聚物的两个或假设干个不同单体的均聚物玻璃化温度之间.接枝共聚物、嵌段共聚物和两种均聚物的共混物, 一般都有两个或多个玻璃化温度值.⑸交联的影响当交联度不大时, T g 变化不大;当交联度增大时, T g 随之增大.⑹增塑剂的影响增塑作用:为便于成型加工或改良高聚物的某些物理力学性能, 常常在高聚物中参加某些低分子物质, 以降低高聚物的玻璃化温度和增加其流动.增塑剂:通常参加的低分子物质多数是沸点高,能与高聚物混溶的低分子液体物质.⑺外界条件的影响外力作用的时间、升温的速率对玻璃化温度都有影响.3 .玻璃化温度的测定方法主要依据:高聚物在发生玻璃化转变的同时, 高聚物的物性参数发生变化.最常用的方法有:热-机械曲线法、膨胀计法、电性能测试法、差热分析法和动态力学法等.二、熔点1 . 熔点的定义与应用熔点:是在平衡状态下晶体完全消失的温度,用T m表示.应用:对于晶态高聚物的塑料和纤维来说, T m是它们的最高使用温度,又是它们的耐热温度,还是这类高聚物成型加工的最低温度.2 . 小分子结晶物质的熔融过程与晶态高聚物其结晶的熔融过程熔融过程:从晶相到液相的转变过程.小分子结晶熔融时:热力学函数有突变〔线型为折线〕;熔化的温度范围窄〔T m± 0.1 ℃〕;熔点与两相的含量无关.晶态高聚物熔融时:快速升温线型渐进线〔非折线〕;熔化的温度范围宽;且熔点与两相的含量有关.即晶态高聚物的熔融过程与小分子结晶的熔融过程只有程度上差异, 而无本质上的不同.最大的差异是:小分子结晶的熔点无记忆性〔与结晶的过程无关〕,而晶态高聚物的熔点有记忆性〔与结晶的过程有关〕.3 .影响熔点的因素T m是结晶高聚物的最高使用温度,所以T m越高,对使用越有利.在平衡熔点时,F=A HI- TA S= 0熔点T m 为:T m= △H/AS可知:AH越大或△ S越小,那么高聚物的T m越高.:对结晶性高聚物进行高度拉伸,以使拉伸的结晶完全,进而提升熔点.4 .熔点的测定方法T m测定方法:与玻璃化温度的测定方法相同.三、粘流温度1 . 粘流温度的定义与应用定义:是非晶态高聚物熔化后发生粘性流动的温度, 又是非晶态高聚物从高弹态向粘流态的转变温度,用T f 表示.应用:是这类高聚物成型加工的最低温度. 这类高聚物材料只有当发生粘性流动时,才可能随意改变其形状.因此,粘流温度的上下,对高聚物材料的成型加工有很重要的意义.粘流温度越高越不易加工.2 .影响粘流温度的因素柔性越大, T f 越低;反之,刚性越大, T f 越高.高聚物的平均相对分子质量越大, 分子间内磨擦越大, 大分子的相对位移越难,T f越高.3 .粘流温度的测定方法热- 机械曲线法、差热分析法注意:T f要作为加工温度的参考温度时, 测定时的压力与加工条件越接近越好.四、软化温度定义:是在某一指定的应力及条件下〔如试样的大小、升温速度、施加外力的方式等〕 ,高聚物试样到达一定形变数值时的温度,一般用Ts 表示.应用:是生产部门产品质量限制、塑料成型加工和应用的一个参数.表示方法:1. 马丁耐热温度2.维卡耐热温度3.弯曲负荷热变形温度〔简称热变形温度〕五、热分解温度定义:是高聚物材料开始发生交联、降解等化学变化的温度,用T d 表示.应用:显示了高聚物材料成型加工不能超过的温度.测定:采用差热分析、热失重、热-机械曲线等方法.六、脆化温度定义:是指材料在受强力作用时,从韧性断裂转为脆性断裂时的温度,用T b第三节高聚物的力学性能力学性质:研究物体受力作用与形变的关系.学习目的:为高聚物材料的成型加工打下良好的根底.、材料的力学概念根本概念:外力、内力、形变、应力、应变、强度、泊松比、模量、柔量、抗张强度、抗弯强度、抗冲击强度、硬度、回弹性、韧性、疲劳等.力学概念定义外力对材料所施加的、使材料以形变的力,一般又称为负荷〔如拉力、压力等〕内力材料为对抗外力、使材料保持原状所具有的力形变材料的变形值,4对形变用^ x、Ay> Az表示;相对形变用△ x/x、△ y/y、△ z/z 表小应力单位面积所受的力,一般用b表示应变在应力作用下,单位长度〔单位面积或单位体积〕所发生的形变,称为应变.一般用丫或e表不强度在一定条件下,材料所能忍受的最大应力称为强度. 常用单位为MPa泊松比横向应变与纵向应变之比, 在理想的情况下:£y= £z=- v s x, V称为泊松比模量模量是引起单位应变所需要的应力,一般用E表示常见:拉伸模量、压缩模量、切变模量、弯曲模量意义:反映高聚物材料的硬性或刚性, E大,刚性大.柔里模量的倒数抗张强度又称拉伸强度,是在规定的温度、湿度和加载速度F, 在试样上沿轴向施加拉力直到试样被拉断为止, 断裂前试样所承受的最大载荷与试样截面之比称为抗张强度抗弯强度又称绕曲强度,是在规定条件下对标准试样施加静弯曲力矩,取试样断裂前的最大载荷抗冲击强度又称抗冲强度或冲击强度,是衡量材料韧性的一种指标.一般是指试样受冲击载荷而破裂时单体面积所吸收的能量测试方法:高速拉伸法、摆锤法、落重法.硬度硬度表示材料反抗其它较硬物体压入的性质, 是材料在一定条件下的软硬程度的性质指标, 用以反映材料承受应力而/、发生形状变化的水平回弹回弹性表示材料吸收能量而不发生永久形变的水平, 一般用回弹能表示韧性韧性表示材料吸收能量并发生较大的永久形变, 但不产生断裂的水平,可用〔T - e曲线下面整个面积来表示.疲劳次-种材料受到屡次形变时, 它的性质会发生改变.在屡次重复施加应力和应变后,力学性质的衰减或损坏通称为疲劳二、等速拉伸及应力-应变曲线1 .非晶态高聚物的应力-应变曲线A点〔弹性极限〕:b - e关系服从虎克定律Y点〔屈服点〕:即使应力不再增加,材料仍能继续发生一定的伸长,B点〔断裂点〕在拉伸过程中,高分子链的运动分别经过三种情况.⑴弹性形变虎克定律:Ee,斜率E为弹性模量.此段主要是由分子链内键长、键角的变化所导致的普弹性能, 有时也包括高弹性形变.⑵强迫高弹形变对常温处于玻璃态的高聚物, 本来链段运动是不能发生的, 之所以能发生,是由于施以强力,强迫它运动,如果除去外力,由于高聚物本身处于玻璃态,在无外力时,链段不能运动,因而高弹形变被固定下来,成为“永久形变〞,因此,屈服强度是反映塑料对抗永久形变的水平.强迫高弹形变可达300 %〜1000 %.这种形变从本质上说是可逆的.但对塑料来说,那么需要加热使温度高于玻璃化温度才有可能消除.⑶粘流形变2 .从材料力学性能曲线形状上,可以把非晶态高聚物的应力-应变曲线大致分为六种:①材料硬而脆,具有高模量及抗张强度, 断裂伸长率很小,受力时呈脆性断裂,可做刚性制品,但不宜受冲击,用于承受静压力的材料. 如酚醛制品(d B^J 50MPa o②材料硬而强,具有高模量及抗张强度, 断裂伸长率亦较小,根本无屈服伸长, 如硬聚氯乙烯.(6)非晶态高聚物的应力-应变曲线③材料强而韧,具有高模量及抗张强度,断裂伸长率较大,有屈服伸长.材料受力时,多属于韧性破坏,受力部位会发白,如聚碳酸酯(b B= 66〜70 MPa E= 2.4 X 104, £B=100%).上述三种,由于强度较大,一般可作工程塑料应用.④材料软而韧,模量低,屈服强度低,断裂伸长率大( 200%〜1000%),断裂强度亦相当高,用于要求形变较大的材料,如硫化橡胶、高压聚乙烯等.⑤材料软而弱,低模量,低抗张强度,但仍有中等的断裂伸长率,如未硫化的天然橡胶,在加工过程中(如汽球成型)需利用这些特性,用吹气胀大到达所需要求的形状后再硫化,成为④类材料.⑥材料弱而脆,一般为低聚物,无做材料的应用价值.可见:强与弱,可以用b B来判断;硬与软,用E的大小来判断;韧与脆,用曲线下面的面积大小来判断.所谓脆性断裂:是指在拉伸时, 未达屈服强度而材料就断裂, 一般断裂伸长小,因而曲线下面积小, 并且断面较平整或有贝壳状;所以韧性的大小可用曲线下面积大小来衡量,它表示材料断裂前所能吸收的最大能量.E 反映单位弹性伸长所需的应力, 表示材料的刚性, 其单位为MPa. 对一般高聚物,E的范围为:橡胶0.1〜1.0 MPa,塑料10〜103MPa ,纤维103〜104MPa , 低分子晶体103〜107 MPa.2.晶态高聚物的应力-应变曲线曲线具有更明显的转折,整个曲线有两个转折点,划分为三段三、影响强度的因素1 . 相对分子质量及分布的影响1/ 当相对分子质量小时,强度随相对分子质量的增高而增大.2/ 当相对分子质量足够大时,强度与相对分子质量大小无关,这种破坏称为分子内破坏机理.2 .低分子掺合物的影响一般会使材料的强度下降.3 .交联、结晶、取向的影响交联:一般有利于强度的提升;过多交联时,也会造成强度下降.结晶:一般情况,结晶使高聚物变硬变脆.取向:对高聚物的所有力学性能都有影响, 最突出的是取向产生各向异性和取向方向的强度.4 .填充物的影响添加活性填料, 如补强剂和增强剂等可以提升分子间的结合力, 预防裂缝的增长. 如炭黑对橡胶的补强作用, 各种类型的增强塑料. 活性二氧化钛对硅橡胶的增强,木粉、棉布、玻璃纤维对热固性塑料的增强等.5 .材料中缺陷的影响缺陷往往用眼睛是看不见的,但对性质特别是对冲击强度的影响是巨大的.四、高聚物的松弛性质〔松弛现象〕1. 松弛现象物体从一种平衡状态过渡到另一种平衡状态的过程.常见:应力松弛和蠕变.(1 〕应力松弛在保持高聚物材料形变一定的情况下, 应力随时间的增长而逐渐减小的现象.(2〕蠕变在一定的应力作用下,形变随时间而开展的现象.能作为结构材料或机械零件作用的工程塑料,均要求蠕变或应力松弛小. 2.蠕变曲线、应力松弛曲线(1 〕蠕变曲线蠕变曲线分为三种类型:①停止型作为工程塑料使用的高分子材料, 为了保证其长期使用的要求, 一般应选用具有这类型蠕变的材料,并且蠕变极限值越小越好.②稳变型曲线相应于常温下处于高弹态的非晶高聚物③增长型曲线相应于常温处于粘流态的非晶态高聚物,发生了粘性流动.(2〕应力松弛曲线两种形式:停止型和减小型. 一般对线型的高聚物来说, 停止型只是减小型的中间阶段, 由于松弛所需的时间长, 在实验时间内未能明显观察到应力进一步减小的情况,作为工程塑料需要这种类型.3 .影响蠕变、应力松弛的因素⑴内因一一指与高聚物本身的化学及物理结构有关的因素凡能使大分子间相互作用增大或使链段长度增大的因素, 都能使蠕变及应力松弛减小.⑵外因一一指与温度、应力、填料、增塑剂等有关的因素①温度温度增高,蠕变速率和数值增大〕而且随着温度的升高, 蠕变从停止型依次向稳变型、增长型转变.②应力应力增大,其效果与温度升高相类似③填充、增强有利于降低材料的蠕变值.④增塑剂随增塑剂的参加,使材料的塑性增加,因此有利于应力松弛及蠕变的开展.4 .松弛时间完成松弛过程所需的时间称为松弛时间.一般用.表示..的定义为:Ae RT式中⑴一一松弛时间;A ⑴一常数;⑴一一重排位垒;R ⑴一气体常数;T ——绝对温度.五、复合材料的力学性质这种材料不但加工方便, 而且能够复合材料是由纯树脂中参加添加剂所组成.满足生产和日常生活上的各种各样要求.制备复合材料的方法:化学共聚方法和物理方法.化学共聚方法在本书第二章节介绍,这里重点介绍物理方法,即增塑、增强、填充及高聚物的共混等.1. 高聚物的增塑作用指能使大分子链的柔性或材料的可塑性增大的作用.增塑作用可分为:内增塑、外增塑和自动增塑三类.〔1 〕内增塑作用是通过改变大分子链的化学结构,以到达增塑的目的.它实际上是化学改性,即通过共聚、大分子反响等化学方法来改变大分子链柔顺性. 这种增塑效果是最稳定,如高抗冲聚苯乙烯.〔2〕外增塑作用在刚性链中参加低分子液体或柔性链的聚合物以到达增塑的目的.①增塑剂:具有该作用的物质称为.②增塑剂的增塑作用:降低高聚物的玻璃化温度和粘流温度③增塑作用的机理:增塑剂起了屏蔽作用和隔离作用.④关于增塑剂的选择原那么:先是增塑剂必须与高聚物材料互溶;其次是不易挥发〔沸点较高〕,能长时间保存在制品中;再有是毒性、颜色、价格等方面.〔3〕自动增塑作用是指非人为参加增塑剂,而是由于某些自动的原因,如高聚物中含有单体、低聚物或混入了杂质、吸收了水分所引起的增塑作用.2.物材料的增强及填充〔1 〕增强作用:高聚物中加一些补强剂或增强剂,使其强度得到不同程度的提升,增强塑料在建筑器材、机器零部件、交通工具、电工零件等各方面获得越来越多的应用热塑性塑料的增强, 为了保持能用注塑机、挤出机成型, 而多使用天然或合成纤维、玻璃纤维、石棉纤维、玻璃微珠、碳纤维等.工业上最常用的是玻璃纤维.经过玻璃纤维增强后的材料与纯树脂相比,具有如下性能:静态强度:如抗张强度、弯曲强度提升2〜4倍;动态强度:耐疲劳性能提升2〜3倍;冲击强度:脆性材料提升2〜3倍,韧性材料那么变化不大;蠕变强度:提升2〜5倍;热变形温度:均有所提升,但幅度不大,为10c〜200 c不等,其中无定型树脂提升的幅度小,结晶高聚物提升的幅度较大.线膨胀系数、成型收缩率及吸水率等均下降.〔2〕填充:在高聚物材料中参加填充剂的过程.填充的目:可以是改性,也可以是单纯的降低本钱.如使用活性填充剂,即填充剂高聚物材料有较强的相互作用, 能使强度提升. 如橡胶中填充炭黑后, 可以提升轮胎的耐磨性和弹性模量, 所以称它为补强剂. 对强度无影响的称为非活性填充剂,如碳酸钙、粘土、木屑等.又如用玻璃纤维填充塑料,称为玻璃纤维增强塑料. 颗粒状活性填充剂具有交联作用, 可以提升材料的强度和刚性.3.高聚物材料的共混改性将结构不同的均聚物、共聚物甚至将相对分子质量不同的高聚物, 通过一定的方法相互掺混,以获得材料的某些特定性能的方法.第四节高聚物的粘流特性一、高聚物的流变性指高聚物有流动与形变的性能.从高聚物材料受力后形变与时间的关系, 以及应力和应变的关系来看, 可以把材料形变分为九种类型.二、影响流变性的因素1 . 相对分子质量及其分布的影响粘度是液体流动时内磨擦力的表征. 对刚性高分子链, 其链段的尺寸趋近于整个大分子链的大小, 因而平均相对分子质量越大, 流动时的有效体积越大, 即粘度越大,流动性越小.2 .温度的影响3 .应力的影响三、高聚物熔体流动中的弹性效应指高聚物熔体〔粘弹性流体〕在压力下从模具的模口被挤出时, 料流立即膨胀,所得挤出物的横截面大于模口截面积的现象〔又称出口膨胀现象〕.是一种弹性滞后效应,也称巴拉斯效应.四、熔融粘度测定测定方法:毛细管粘度计法〔包括熔融指数测定仪等〕是测量在一定压力下液体自毛细管中流出来的速率;旋转式粘度法〔包括悬锤- 杯式、锥-板式或门尼粘度计等〕, 是使一个圆体〔筒、锥或盘〕旋转在高聚物液体中.第六节高聚物溶液与相对分子质量在高聚物的实际生产中,高聚物的分级、相对分子质量的测定、溶液的纺丝、油漆、絮凝剂、分散剂、泥浆处理剂等都是在溶液状态下进行的,而高聚物溶液的性质又与高分子的形态、大分子的相互作用、高分子链的支化程度密切相关, 因此对高聚物溶液的研究无论在理论和实际应用上均有重要意义.高聚物溶液:高聚物以分子状态分散在溶剂中形成的均相体系. 是真溶液, 由于高聚物本身特征,如分子量很大、多分散性,高分子几何形状有线型、支链型和交联型以及聚集态有晶态、非晶态,因此,高聚物比低分子溶解复杂的多.、高聚物的溶解1 .非晶态高聚物的溶解现象:糖+水一溶解快苯乙烯+苯一溶解慢,并且要先溶胀形成透明胶冻物质,然后溶解.分析:小分子溶解水糖分子大小相似,运动速度为相同数量级,双向扩散相互钻入对方的空穴.高聚物溶解苯乙烯苯分子大小相差悬殊,小分子几个A,大分子100—1000A,运动速度相差大,只能单方向扩散.大分子扩散速度比小分子扩散速度低, 所以小分子能迅速向高聚物扩散,故高聚物溶解之前体积膨胀(溶胀),相互扩散,形成溶液(溶解).(1)溶解过程三个阶段:三种运动单元:溶剂分子、高分子链段和整个高分子链前期:局部链段松动中期:大局部链段松动,溶剂分子不断深入高分子链之间, 使高聚物的体积膨胀,即溶胀,但大分子还不能脱出整体进入溶剂中.后期:大分子脱离整体进入溶剂中,形成均相溶液.(2)溶解的过程图示溶解〞a)非晶高聚物的溶解过程(a)线型非晶高聚物的溶解过程条件:溶剂足够特点:慢,无限溶胀(溶解)体系运动单元:溶剂分子、链段、大分子链(b)网状高聚物的溶胀条件:溶剂足够特点:慢,有限溶胀(不溶解) 体系运动单元:溶剂分子、链段总之,非晶高聚物的溶解过程中,最关键的步骤是溶胀( swelling).2 .结晶高聚物的溶解—聚而得——极性结晶高聚物分两类'加聚而得——非极性结晶高聚物(1)非极性结晶高聚物的溶解首先要加热到熔点附近,使结晶熔化,成为无定型的液态, 再按上述溶解过程溶解.如低压聚乙烯在四氢蔡中要在 120C,全同立构或间同立构聚丙烯在十氢蔡中要在130c 才能很好溶解.(2)极性结晶高聚物要选择适宜的强极性溶剂,在常温下就可以溶解.运动单元:溶剂分子溶剂分子 溶剂分子 局部链段落(b)大局部链段所有链段。
《塑料成型加工基础》单元电子教材高分子的特征温度——熔点一、熔点晶态高聚物的熔点是在平衡状态下晶体完全消失的温度。
一般用T m表示。
对于晶态高聚物的塑料和纤维来说,T m是它们的最高使用温度,又是它们的耐热温度,还是这类高聚物成型加工的最低温度。
低分子晶体的熔融过程晶态高聚物的熔融过程晶态高聚物熔融的特点:(1)晶态高聚物熔融时比容-温度关系是边熔融边升温,突变不明显。
(2)存在熔限,即较宽的熔融温度范围的3~4K。
高分子结晶熔融过程中熔限的宽窄同高分子结晶的形成条件密切相关。
晶态聚合物边熔融边升温的主要原因主要是由于结晶聚合物中含有完善程度不同的晶体的缘故。
结晶时随着温度的下降,黏度上升,分子链活动能力减小,还来不及作充分的位置调整,这样各个不同阶段结晶状态同时并存,当熔化不完善的晶体(分子链堆不紧密)将在较低的温度下熔融,而完善的晶体则在较高温度下才能熔融,所以在通常升温条件下便出现较宽的熔融温度范围。
二、影响熔点的因素因为熔点是结晶高聚物的最高使用温度,所以熔点越高,对使用越有利。
因此,我们通过分析影响熔点的因素,找到合适的途径提高熔点。
在平衡熔点时,高聚物的晶相与非晶相达到热力学平衡,△F=0,即△F=△H-T△S=0所以其熔点为:T m=△H/△S式中,△H——1摩尔重复结构单元的熔化热;△S——1摩尔重复结构单元的熔化熵。
由此可知,△H越大或△S越小,则高聚物物的熔点越高。
△H与分子间作用力的强弱有关,若在高分子主链中或侧基上引入极性基团,或在大分子间形成氢键,均能增大分子间的作用力,进而,提高△H。
△S与晶体熔化后分子的混乱程度有关,进而与分子链的柔性有关。
柔性越好,晶体熔化后分子链的混乱程度就越大,因此其熔点就越低。
当主链引入苯环时,则柔性下降,刚性增加,因此使熔点升高。
另外一种工业上常用的方法,对结晶性高聚物进行高度拉伸,以使结晶完全,进而提高熔点。
三、熔点的测定方法熔点的测定方法基本上与玻璃化温度的测定方法相同。
第五章 高聚物的物理性能第一节 高聚物的物理状态高聚物的聚集态结构,根据链结构的规整性和能否结晶可分为两类: 结晶性高聚物(有规则排列)非结晶性高聚物(无规则排列)链段运动——使高聚物具有高弹性高聚物热运动具有两重性整个分子链运动——使高聚物象液体一样具有粘流性热-机械曲线——形变-温度曲线:表示高聚物材料在一定负荷下,形变大小与温度的关系曲线。
按高聚物的结构可以分为:线型非晶高聚物形变-温度曲线结晶态高聚物形变-温度曲线 其他类型的形变-温度曲线三种一、线型非晶态高聚物的物理状态1.形变-温度曲线A B C D ET b T g 温度(℃) T fT b -脆化温度;T g -玻璃化温度;T f -粘流温度可分为五个区A 区(玻璃态):内部结构类似玻璃,大分子不能运动,链段也不能运形变(%)动,在除去外力后,形变马上消失而恢复原状,可逆形变称为普弹性形变。
C 区(高弹态或橡胶态):除了普弹形变外,主要发生了大分子的链段位移(取向)运动。
但整个大分子间并未发生相对位移,形变也可以消除,所以是可逆的弹性形变。
E 区(粘流态或塑化态):当施加负荷时,高聚物象粘性液体一样,发生分子粘性流动,大分子能运动,链段也能运动,形变不能自动全部消除,这种不可逆特性,称为可塑性。
B 区和D 区:为过渡区。
其性质介于前后两种状态之间。
玻璃态物理力学三态高弹态 (是一般非晶态高聚物所共有的)粘流态2.非晶态高聚物三种物理状态的力学行为特征和形变机理3.三态之间的转变随温度的变化而逐渐变化过程 玻璃态 ⇔高弹态⇔ 粘流态 4.注意问题1/ T g 是大分子链段能运动的最低温度,高弹态的出现是链段运动的产物。
2/ T g 与柔性的关系:柔性大,T g 低,反之。
刚性大,T g 高。
3/ T g 与T f 的使用价值T g 是塑料、纤维的最高使用温度T f 是橡胶的最低使用温度,也是高聚物成型加工温度。
5.线型非晶态高聚物的物理力学状态与相对分子质量的关系不同相对分子质量的聚苯乙烯的热-机械曲线二、结晶态高聚物的物理状态晶态高聚物的形变-温度曲线 1-一般相对分子质量 2-相对分子质量很大1/ 结晶态高聚物按成型工艺条件的不同可以处于晶态和非晶态。
第一章高分子链的结构全同立构:高分子链全部由一种旋光异构单元键接而成间同立构:高分子链由两种旋光异构单元交替键接而成构型:分子中由化学键所固定的原子在空间的几何排列,这种排列是热力学稳定的,要改变构型必需经过化学键的断裂与重组分子构造(Architecture):指聚合物分子的各种形状,一般高分子链的形状为线形,还有支化或交联结构的高分子链,支化高分子根据支链的长短可以分为短支链支化和长支链支化两种类型共聚物的序列结构:是指共聚物根据单体的连接方式不同所形成的结构,共聚物的序列结构分为四类:无规共聚物、嵌段共聚物、交替共聚物、接枝共聚物接枝共聚物:由两种或多种单体经接枝共聚而成的产物,兼有主链和支链的性能。
嵌段共聚物(block copolymer):又称镶嵌共聚物,是将两种或两种以上性质不同的聚合物链段连在一起制备而成的一种特殊聚合物。
环形聚合物:它的所有结构单元在物理性质和化学性质上都是等同的超支化聚合物:是在聚合物科学领域引起人们广泛兴趣的一种具有特殊大分子结构的聚合物构象:由于σ单键内旋转而产生的分子在空间的不同形态。
链段:高分子链上划分出的可以任意取向的最小单元或高分子链上能够独立运动的最小单元称为链段。
链柔性:是指高分子链在绕单键内旋转自由度,内旋转可导致高分子链构象的变化,因为伴随着状态熵增大,自发地趋向于蜷曲状态的特性。
近程相互作用:是指同一条链上的原子或基团之间,沿着链的方向,因为距离相近而产生相互作用远程相互作用:因柔性高分子链弯曲所导致的沿分子链远距离的原子或基团之间的空间相互作用。
远程相互作用可表现为斥力或引力,无论是斥力还是引力都使内旋转受阻,构想数减少,柔性下降,末端距变大。
自由连接链:假定分子是由足够多的不占体积的化学键自由结合而成,内旋转时没有键角限制和位垒障碍,其中每个键在任何方向取向的几率都相同。
(极端理想化假设)自由旋转链:假定链中每一个键都可以在键角所允许的方向自由转动,不考虑空间位阻对转动的影响等效自由连接链:若干个键组成的一链段算作一个独立的单元,称之为“链段”,链段间自由结合,无规取向,这种链的均方末端距与自由连接链的计算方式等效。