简化基因组测序
- 格式:pdf
- 大小:2.50 MB
- 文档页数:29

基于简化基因组测序的大黄鱼耐高温性状全基因组关联分析陈小明;李佳凯;王志勇;蔡明夷;韩芳;刘贤德【期刊名称】《水生生物学报》【年(卷),期】2017(041)004【摘要】利用Illumina HiSeqTM 2500测序平台,对通过高温胁迫实验筛选得到的20尾耐高温和20尾不耐高温的大黄鱼(Larimichthys crocea)进行了简化基因组测序(SLAF-seq),每个样本的平均测序深度达到10.26×,共获得419211个高质量的群体单核苷酸多态性(SNP)位点.利用TASSEL软件的混合线性模型(MLM)进行全基因组关联分析(GWAS),共筛选到38个与大黄鱼耐高温性状显著相关的SNP 位点(P<2.39E–08).利用BLAST程序定位每个SNP位点在大黄鱼基因组中的位置,并分析其周围的功能基因.结果在38个SNPs附近共找到26个已知的功能基因,这些基因主要与细胞转录、代谢、免疫等功能相关.研究结果可为下一步大黄鱼耐高温分子机制解析及耐高温品种的选育提供参考.%Twenty thermal-tolerant and twenty thermal-sensitive individuals ofLarimichthys crocea were sequenced using specific-locus amplified fragment (SLAF-seq) technology based on Illumina HiSeqTM2500 platform. 419211 SN-Ps were identified with an average read depth of 10.26× for each sample. Thirty-eight SNPs(P<2.39E–08) signifi-cantly related with thermal tolerance trait were identified according to association analysis. The SNP locations in large yellow croaker genome were identified using BLAST program, and functional genes around SNP were annotated. Twenty-six genes with known functions were discovered around 38 SNPs, which mainly regulatecell transcription, metabolism and immunity. These results provide basic information to analyze thermal-tolerant molecular mechanism and develop thermal-tolerant lines ofLarimichthys crocea in the future.【总页数】6页(P735-740)【作者】陈小明;李佳凯;王志勇;蔡明夷;韩芳;刘贤德【作者单位】集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门361021;集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门 361021;集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门 361021;集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门 361021;集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门 361021;集美大学水产学院,农业部东海海水健康养殖重点实验室,厦门 361021【正文语种】中文【中图分类】Q344+.1【相关文献】1.基于简化基因组测序技术和基因芯片技术比较研究黄羽肉鸡基因组选择 [J], 刘天飞;罗成龙;王艳;周广源;马杰;舒鼎铭;苏国生;瞿浩2.基于MAGIC群体的水稻抽穗期和产量相关性状全基因组关联分析 [J], 魏秀彩;李小湘;刘金栋;刘利成;黎用朝;潘孝武;董铮;刘文强;熊海波;闵军3.基于50K SNP芯片技术对金华猪和嵊县花猪繁殖性状的全基因组关联分析 [J], 蔡薇;罗才玉;项云;章啸君;徐宁迎;郭晓令4.基于简化基因组测序的红罗非鱼低温体色变异全基因组关联分析 [J], 徐鸿飞;朱华平;陈诏;黄彩林;袁宗伟;赵何勇;李华;杨宾兰;周大颜;苏换换5.基于SNP标记的小麦籽粒性状全基因组关联分析 [J], 张芳;任毅;曹俊梅;李法计;夏先春;耿洪伟因版权原因,仅展示原文概要,查看原文内容请购买。
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS (Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
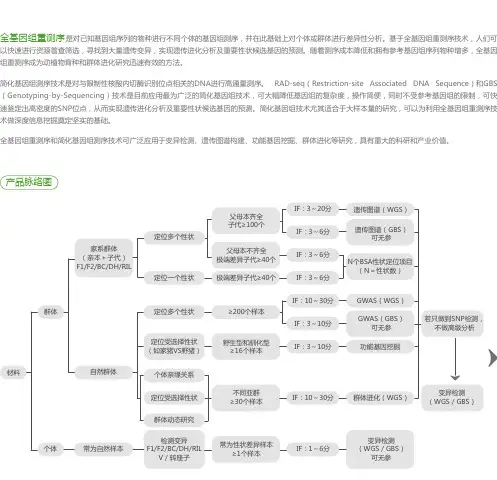
产品脉络图。

doi: 10.7541/2017.91基于简化基因组测序的大黄鱼耐高温性状全基因组关联分析陈小明李佳凯王志勇蔡明夷韩芳刘贤德(集美大学水产学院, 农业部东海海水健康养殖重点实验室, 厦门 361021)摘要: 利用Illumina HiSeq TM 2500测序平台, 对通过高温胁迫实验筛选得到的20尾耐高温和20尾不耐高温的大黄鱼(Larimichthys crocea)进行了简化基因组测序(SLAF-seq), 每个样本的平均测序深度达到10.26×, 共获得419211个高质量的群体单核苷酸多态性(SNP)位点。
利用TASSEL软件的混合线性模型(MLM)进行全基因组关联分析(GWAS), 共筛选到38个与大黄鱼耐高温性状显著相关的SNP位点(P<2.39E–08)。
利用BLAST程序定位每个SNP位点在大黄鱼基因组中的位置, 并分析其周围的功能基因。
结果在38个SNPs附近共找到26个已知的功能基因, 这些基因主要与细胞转录、代谢、免疫等功能相关。
研究结果可为下一步大黄鱼耐高温分子机制解析及耐高温品种的选育提供参考。
关键词: 大黄鱼; 高温胁迫; 简化基因组测序; 单核苷酸多态性; 全基因组关联分析中图分类号: Q344+.1 文献标识码: A 文章编号: 1000-3207(2017)04-0735-06大黄鱼(Larimichthys crocea)是我国重要的海洋经济鱼类, 在自然海区分布于30—60 m水深, 适应温度在10—32℃, 最适生长温度在18—25℃[1, 2]。
当前, 大黄鱼的养殖模式仍以浅海网箱养殖为主,网箱深度为4—6 m。
由于水深较浅, 夏季大黄鱼处在(或接近)其可耐受高温的时间较长, 持续高温会导致大黄鱼生长减缓、抗病力下降, 加上病原生物感染, 常常引发大黄鱼大量发病死亡。
因此, 开展大黄鱼耐高温选育的研究, 对提高养殖大黄鱼的度夏成活率具有重要的参考意义。

#流程大放送#WGBS和RRBS测序分析流程介绍WGBS全称Whole Genome Bisulfite Seuqneicng,即全基因组重亚硫酸盐测序。
该方法通过Bisulfite处理,将原基因组中未发生甲基化的C碱基转换成U的同时,保留所有甲基化C 的碱基不发生转变,从而帮助科研人员识别发生甲基化的CpG位点。
该种测序技术适用于绘制单碱基分辨率的全基因组DNA甲基化图谱。
RRBS全称Reduced Representation Bisulfite Sequencing,即简化代表性重亚硫酸盐测序。
该方法在Bisulfite处理前,使用MspI(该酶的酶切位点为CCGG)酶切对样本进行处理,去除低CG含量DNA片段,从而使用较小的数据量富集到尽可能多的包含CpG位点的DNA片段。
相比于WGBS技术,RRBS是一种准确、高效且经济的DNA甲基化研究方法,通过酶切,并进行Bisulfite测序,该方法在保证DNA甲基化状态检测的高分辨率的同时提升测序数据的高利用率。
该项技术可用于以下研究1、处于特定时期或特定处理条件下的样本中,研究样本中染色体高精度DNA甲基化模式;2、比较不同细胞、组织、样本间的高精度DNA甲基化修饰模式的差异;3、疾病样本中,与疾病发生发展相关的高精度DNA甲基化表观遗传机理研究和相关高精度DNA甲基化位点分子标志的探索性研究。
数据处理和分析流程图分析结果示例图片展示示例图1 样本中各区域DNA甲基化水平信息统计和样本间差异DNA甲基化分析结果展示[1]示例图2 差异DNA甲基化区域内转录因子基序识别[1]示例图3 DNA甲基化水平变化与基因表达水平变化的关联性分析[1]示例图来源文献[1]. Ng, C.W., et al., Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci U S A, 2013. 110(6): p. 2354-9.。



简化基因组甲基化测序
简化基因组甲基化测序是一种新兴的技术,可用于确定某个细胞或组
织的基因组DNA中哪些部位被甲基化。
甲基化是一种常见的基因表达调控方式,常常用于关闭或压制基因表达,从而影响细胞的功能。
简化基因组甲基化测序通常基于第二代测序技术,可以比较容易地确
定哪些基因组部位与可编码蛋白质相关,以及哪些部位在某些特定细
胞或组织类型中被甲基化。
这项技术可以帮助科学家更好地理解基因
调控的细节,同时为许多人类疾病的研究提供了极为重要的基础。
但是,简化基因组甲基化测序也存在着一些挑战。
首先,一些技术问
题如样品制备和DNA富集可能会影响结果的准确性。
此外,由于甲基化通常发生在基因组中的小的CpG岛上,这对于大部分组织来说并不是非常普遍,所以简化基因组甲基化测序结果并不能覆盖所有的基因
组区域。
同时,受限于设备和分析算法的限制,数据规模可能无法满
足一些研究所需。
虽然简化基因组甲基化测序存在一定的局限性,但它还是一项非常有
价值的技术,可以帮助科学家更好地理解基因组和表观遗传学的作用。
未来,我们可以更为准确地理解某些人类疾病的发生机制,并且在疾
病治疗中使用这些知识,以促进更好的健康和预防。



基于简化基因组测序评估陇南山羊群体遗传多样性和群体结构马克岩;刘占经;白雅琴;马友记【期刊名称】《中国畜禽种业》【年(卷),期】2024(20)4【摘要】该试验旨在利用简化基因组测序(specific-locus amplified fragment sequencing, SLAF-seq)分析陇南山羊群体遗传多样和群体结构,为陇南山羊后续保种计划的制订提供依据。
该研究对50只陇南山羊(公、母各25只)进行全基因组SNP检测;计算观测杂合度(Ho)、期望杂合度(He)、多态信息含量(PIC)、香浓维纳指数(SHI)、基因多样性指数(Nei)及次要等位基因频率(MAF)等6个指标进行遗传多样性评估。
结果表明,50只陇南山羊共检测到655514个SNPs位点。
陇南山羊的Ho、 He、 PIC、 SHI、 Nei及MAF指标值分别为0.193、0.286、0.236、 0.449、 0.290、 0.200,说明该群体遗传多样性较低。
陇南山羊群体LD 值较低,衰退速度较快,说明该群体并未受到过强烈的人工选择压力。
此外,PCA与系统发育树结果均表明陇南山羊群体内部出现分化,可分为2个大的亚群。
陇南山羊群体平均IBS遗传距离为0.7391,结合亲缘关系G矩阵热图,表明陇南山羊群体大部分个体亲缘关系较远。
50只陇南山羊个体共检测到47206个ROH,平均ROH 长度37.86 Mb,基于ROH的近交系数FROH范围为0.0535~0.2574,平均FROH 值为0.1472,说明陇南山羊群体存在近交风险。
可见,陇南山羊内部存在分化,可分为2个大的亚群,陇南山羊群体遗传多样性较低,部分个体间存在较大的近交风险,可能需要采取保种措施保护陇南山羊遗传资源。
【总页数】10页(P8-17)【作者】马克岩;刘占经;白雅琴;马友记【作者单位】甘肃农业大学动物科学技术学院;天祝县动物疫病预防控制中心;甘肃省畜牧技术推广总站【正文语种】中文【中图分类】S813.9【相关文献】1.基于2b-RAD简化基因组测序的半滑舌鳎群体遗传多样性分析2.基于2b-RAD 简化基因组测序的三门湾海域3种优势鱼类群体遗传多样性分析3.基于重测序数据的昌都黑山羊遗传多样性及群体结构分析4.基于简化基因组测序的永登七山羊遗传多样性分析5.基于低深度全基因组测序分析内江猪群体结构和遗传多样性因版权原因,仅展示原文概要,查看原文内容请购买。

专利名称:一种基于简化基因组测序和SNP次等位基因频率的非杂交后代鉴定方法
专利类型:发明专利
发明人:刘有春,袁兴福,刘成,王升,张舵,魏鑫,刘修丽,孙斌,王宏光,杨玉春,高树清
申请号:CN202010736451.7
申请日:20200728
公开号:CN111826429A
公开日:
20201027
专利内容由知识产权出版社提供
摘要:本发明提供了一种基于简化基因组测序和SNP次等位基因频率的非杂交后代鉴定方法,涉及杂交后代鉴定技术领域;所述鉴定方法基于参考基因组,利用SNP次等位基因频率(MAF)数据集,采用遗传关系分析和个体特有的稀有等位变异分析方法,从不同角度反映群体子代间的遗传关系,进而通过箱图直观反映离群个体,确定为非杂交后代,该方法鉴定的非杂交后代与基于双亲纯合显性SNP位点的验证结果一致,因此本发明所述鉴定方法可简单、有效地筛除杂交群体中的非杂交后代,对植物新品种选育及遗传分析、图谱构建、性状定位等研究具有重要意义。
申请人:辽宁省果树科学研究所
地址:115000 辽宁省营口市鲅鱼圈区铁东街
国籍:CN
代理机构:北京高沃律师事务所
代理人:董大媛
更多信息请下载全文后查看。
专利名称:一种基因组简化与二代测序SNP复合检测体系和检测方法
专利类型:发明专利
发明人:李泽卿
申请号:CN201810035165.0
申请日:20180115
公开号:CN108148899A
公开日:
20180612
专利内容由知识产权出版社提供
摘要:本发明公开了一种基因组简化与二代测序SNP复合检测体系和检测方法,包括如下步骤:将形成的扩增产物内添加底物,从而能够合成第一个碱基,并清除所有游离的碱基,在测序时,选取PTP平板,且PTP平板表面含有160万个光纤孔,光纤孔内载有化学发光反应所需的各种酶以及底物,然后将碱基依照T、A、C、G的顺序依次循环进入PTP平板,假如发生碱基配对,会释放焦磷酸,焦磷酸在酶的作用下,形成氧化荧光素,同时释放光信号,实时利用高灵敏度CCD捕获,碱基和PTP平板进行配对,然后捕获到一分子的光信号,由此一一对应,能够准确、快速的确定待测模板的碱基序列。
本发明能够对基因组进行准确的测序。
申请人:武汉爱基百客生物科技有限公司
地址:430000 湖北省武汉市东湖高新区高新大道666号光谷生物城创新园C6栋
国籍:CN
代理机构:上海精晟知识产权代理有限公司
代理人:冯子玲
更多信息请下载全文后查看。
一、名词解释1.系统发育树(phylogenetic tree,又称evolutionary tree进化树):是描述群体间进化顺序的分支图或树,表示群体间的进化关系。
2.主成分分析(PCA):是指将多指标化为少数几个综合指标的一种统计分析方法,能够反映原始变量的绝大部分信息。
3.群体结构:是指一个群体内部的基因频率在不同子群体之间存在着系统性的差异。
二、填空题1.我们公司现有的两种简化基因组测序技术分别是RAD和dd-GBS。
2.简化基因组的主要应用有SNP标记的开发、遗传图谱的构建、群体遗传学分析和QTL 分析。
3.目前用于做RAD测序数据的SNP calling的软件是Stacks。
4.遗传图谱中的遗传距离用厘摩(cM)来表示,1 cM的大小大致符合1%的重组率。
三、选择题1.在构建遗传图谱的时候,通常推荐样本数量至少在B个以上。
A. 50B. 100C. 150D. 2002.遗传图谱是指基因或者DNA标记在染色体上以A表示相对位置的图。
A. 遗传距离B. 物理距离3.常见的暂时性分离群体有A和B;常见的永久性分离群体有C和D。
A. F2B. BC1C. RILD. DH4.为了达到彼此相当的作图精度,所需的群体大小顺序为A>C>B≈D。
A. F2B. BC1C. RILD. DH5.我们公司目前的测序平台有(多选):A. Hiseq2000B. Hiseq2500C. Hiseq4000四、问答题1.RAD 技术的主要流程包括哪几个方面?抽提DNA,质检,建库,测序2.RAD 技术有什么特点和优势?特点:(1)通过酶切作用对基因组特定区域进行测序;(2)反映部分基因组序列结构(变异)信息。
优势:(1)测序量低,价格便宜;(2)数据利用率高,性价比高;(3)实验操作简单;(4)能够构建高密度的分子图谱;(5)不依赖参考基因组,物种适用范围广。
3.RAD 技术和 dd-GBS 技术的主要区别是什么?dd-GBS 技术不对 DNA 片段打断,不需要挖胶和纯化,实验周期比较短。
基于酶切的简化基因组测序(RAD)
1. 对于群体进化研究,个体的混样策略是怎样的?
答:如果更关注群体间的遗传多样性差异,希望消除群体内个体遗传差异带来的干扰,可以采用群体内个体混样的策略,我们会进一步在生物信息分析流程中采用优化的群体内SNP纠错与过滤程序达到分析目的。
2. 我们的RAD-Seq建库测序策略是怎样的?
答:建库策略选择基于综合考虑序列碱基质量、测序成本和物种基因组情况,主要有Single‐end 50 bp和Pair‐end 90 bp。
对于有参考基因组的物种,我们建议采用PE90文库测序。
对于无参考基因组的物种,则建议采用SE50文库测序。
3. RAD-Seq测序结果受哪些因素影响?测序成功的标准是什么?
答:设备、耗材、操作问题等方面都会对测序的结果造成影响。
测序成功标准包括:reads 数达标,错误率低,Q20高等。
基于简化基因组测序技术的油茶SNP标记开发及指纹图谱构建廖宏泽;孙曼曼;黄小娟;郝丙青;孙佳星;江泽鹏;王东雪;刘凯【期刊名称】《中南林业科技大学学报》【年(卷),期】2024(44)4【摘要】【目的】基于简化基因组,挖掘油茶单核苷酸多态性(SNP)位点,筛选可用于油茶种质鉴定的简化SNP组合位点,构建SNP指纹图谱,建立一种快速准确鉴定广西油茶主栽良种苗木的SNP分子标记方法。
【方法】以12个油茶无性系种质的两个重复共24份油茶标准样本为材料,采用ddRADseq流程进行文库构建,使用BWA将过滤后的测序数据比对到已发布的南荣油茶Camellia oleiferavar.“Nanyongensis”参考基因组上,利用GATK进行SNP位点筛选,ANNOVAR 软件进行SNP位点注释,STRUCTURE软件进行群体结果分析,使用PLINK进行主成分分析;利用R语言,使用条件随机筛选法(CRS),筛选能够区分出油茶种质的最简SNP组合,绘制指纹图谱。
【结果】测序数据质量良好,可用于SNP分子标记位点的开发筛选。
与参考基因组比对后,共获得622064个为SNP标记位点,其中无基因型缺失的SNP位点40147个,多态性信息含量(PIC)大于0.35的SNP位点2094个,前后60 bp碱基保守无变异的SNP位点共184个。
最终筛选出可以将12个油茶无性系种质区分开的15个核心SNP标记位点组合,并以此绘制出SNP 指纹图谱。
【结论】基于简化基因组,建立了广西主要栽培油茶种质的SNP标记开发和指纹图谱绘制的方法,为苗期油茶种质的快速准确鉴定提供了理论依据和技术指导,助力规范油茶苗木市场,促进油茶产业健康发展。
【总页数】10页(P128-137)【作者】廖宏泽;孙曼曼;黄小娟;郝丙青;孙佳星;江泽鹏;王东雪;刘凯【作者单位】广西民族大学海洋与生物技术学院;广西壮族自治区林业科学研究院广西特色经济林培育与利用重点实验室【正文语种】中文【中图分类】S794.4【相关文献】1.甘蓝SNP标记开发及主要品种的DNA指纹图谱构建2.基于地黄转录组数据的SNP标记开发与地黄指纹图谱构建3.基于简化基因组测序技术的甘薯HRM分子标记开发及其应用4.基于全基因组SNP构建甘蓝型油菜指纹图谱5.基于KASP技术的SNP标记用于西瓜品种指纹图谱构建和种子纯度检测因版权原因,仅展示原文概要,查看原文内容请购买。
#流程大放送#WGBS和RRBS测序分析流程介绍WGBS全称Whole Genome Bisulfite Seuqneicng,即全基因组重亚硫酸盐测序。
该方法通过Bisulfite处理,将原基因组中未发生甲基化的C碱基转换成U的同时,保留所有甲基化C 的碱基不发生转变,从而帮助科研人员识别发生甲基化的CpG位点。
该种测序技术适用于绘制单碱基分辨率的全基因组DNA甲基化图谱。
RRBS全称Reduced Representation Bisulfite Sequencing,即简化代表性重亚硫酸盐测序。
该方法在Bisulfite处理前,使用MspI(该酶的酶切位点为CCGG)酶切对样本进行处理,去除低CG含量DNA片段,从而使用较小的数据量富集到尽可能多的包含CpG位点的DNA片段。
相比于WGBS技术,RRBS是一种准确、高效且经济的DNA甲基化研究方法,通过酶切,并进行Bisulfite测序,该方法在保证DNA甲基化状态检测的高分辨率的同时提升测序数据的高利用率。
该项技术可用于以下研究1、处于特定时期或特定处理条件下的样本中,研究样本中染色体高精度DNA甲基化模式;2、比较不同细胞、组织、样本间的高精度DNA甲基化修饰模式的差异;3、疾病样本中,与疾病发生发展相关的高精度DNA甲基化表观遗传机理研究和相关高精度DNA甲基化位点分子标志的探索性研究。
数据处理和分析流程图分析结果示例图片展示示例图1 样本中各区域DNA甲基化水平信息统计和样本间差异DNA甲基化分析结果展示[1]示例图2 差异DNA甲基化区域内转录因子基序识别[1]示例图3 DNA甲基化水平变化与基因表达水平变化的关联性分析[1]示例图来源文献[1]. Ng, C.W., et al., Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci U S A, 2013. 110(6): p. 2354-9.。