整理晶体结构解析步骤
- 格式:doc
- 大小:42.00 KB
- 文档页数:7

晶体结构解析
晶体结构解析是通过实验测定晶体的结构,并确定晶体中原子或分子的排列方式、空间坐标和键合情况等信息的过程。
晶体结构解析对于研究材料的物理、化学和生物学性质以及设计新材料具有重要意义。
晶体结构解析的步骤包括:
1. 晶体的培养和选取:选择合适的晶体生长条件,培养出高质量的晶体。
2. X 射线衍射实验:使用 X 射线衍射仪对晶体进行衍射实验,得到衍射图谱。
3. 数据处理和结构因子计算:对衍射图谱进行数据处理,计算结构因子。
4. 结构模型建立:根据结构因子和化学知识,建立晶体的结构模型。
5. 结构精修:通过不断调整结构模型的参数,使其与实验数据相符合,得到最终的晶体结构。
晶体结构解析需要结合化学、物理学和数学等多学科知识,需要专业的实验技能和计算能力。
目前,晶体结构解析已经成为材料科学、化学、生物学等领域的重要研究手段。

晶体结构知识汇总及解题方法技巧一、晶胞中质点得占有率在一个晶胞结构中出现得多个原子,并不就是只为这一个晶胞所独立,而就是为多个晶胞共用,所以每一个晶胞只能按比例分摊。
分摊得根本原则:晶胞任意位置上得原子如果就是被n 个晶胞所共有,则每个晶胞只能分得这个原子得n 1。
立方晶胞,顶点上得粒子占 棱上得粒子占 面上得粒子占 体心得粒子占二、常见晶胞分析1. NaCl 型⑴每个晶胞占有 个Na+, 个Cl-,即 个NaCl 粒子⑵每个Na +周围有 个Cl -,每个Cl -周围有 个Na +,与一个Na +距离最近且相等得Cl-围成得空间构型为 。
每个Na +周围与其最近且距离相等得Na +有 个。
⑶0、585g NaCl 晶体(0、01mol)含有 个晶胞。
⑷若已知Na+与Cl-得最短距离为a cm,则NaCl 晶体得密度为 。
2. CsCl 型⑴在CsCl 晶体中,每个Cs+周围与之最接近得且距离相等得Cs+有 个,每个Cs+周围与之最接近得且距离相等得Cl-有 个。
⑵每个晶胞占有 个CsCl 粒子。
3. 干冰型在干冰晶体中,每个CO2分子周围与之最接近得且距离相等得CO2分子有 个。
每个晶胞中含有 个CO2分子。
4. 金刚石型金刚石得网状结构中,,每个碳原子与其她4个碳原子等距离紧邻,含有由共价键形成得碳原子环,其中最小得环上有6个碳原子,每个碳原子上得任意两个C—C键得夹角都就是109°28′,其中C原子个数:C—C个数= 。
5.石英晶体在二氧化硅晶体中,一个硅原子与4个氧原子形成4个共价键,1个氧原子与2个硅原子形成2个共价键,故Si原子与O原子数目之比为。
实际上,该晶体就是由硅原子与氧原子按1:2得比例组成得立体网状晶体,没有单个分子存在。
在晶体中最小得环为十二元环,每个环占有6个Si原子与6个O原子。
6.石墨晶体结构石墨晶体就是一种混合型晶体,层内存在共价键,层间以范德华力结合,兼具有原子晶体、分子晶体得特征与特性。

晶体结构解析基本步骤1.实验准备阶段:在晶体结构解析之前,首先需要准备精心选择的晶体样品。
由于X射线衍射技术对于晶体品质要求较高,因此必须获得具有高质量的单晶。
通常采用慢结晶法、溶液法或气相法获得单晶。
此外,还需要准备一台高质量的X射线衍射仪。
2.数据收集阶段:在这个阶段,使用X射线衍射仪对晶体样品进行照射。
在衍射仪中,晶体样品会被照射出一系列衍射斑点。
这些斑点的形状和位置与晶体的结构有关。
3.数据处理阶段:在数据处理阶段,需要将从X射线衍射仪中获得的原始数据进行处理。
首先,将原始数据转换成衍射强度和衍射角度的数据。
然后,使用计算机软件对这些数据进行处理和分析,例如标定衍射仪的几何参数,背景的消除,峰的辨识和积分。
4.构建初步模型:在初步模型构建阶段,使用得到的衍射数据来建立原子的初步模型。
这个过程通常是基于一些基本的假设和规则,比如晶胞参数和空间群。
通过将原子位置和晶胞参数进行不断的调整和优化,以找到对衍射数据拟合最佳的结构模型。
5.结构修正阶段:在初步模型构建后,需要对结构进行修正以改善拟合度。
修正的方法包括Rietveld修正、最小二乘法、Patterson法等。
这些方法可以通过比较实验衍射数据和模拟衍射数据来找到原子位置、原子类型和晶胞参数的最佳拟合。
6.结果验证阶段:在得到结构模型后,需要进行结果验证。
这一步通常涉及到测量残差因子和R值,验证得到的结构模型与实验数据的拟合程度。
此外,还可以使用精细调节工具,如法拉第差图和主动位相修正,进一步改善结构的质量。
7.结果分析和报告阶段:最后,通过对解析得到的晶体结构进行分析,得到结构中各个原子的位置、键长、键角及晶胞参数等信息。
然后,将这些结果写入晶体结构解析报告中,并与相应的文献数据进行对比和验证。
总之,晶体结构解析是一个复杂而精细的过程,需要仔细的实验准备、数据处理、构建模型和结果验证等多个步骤。
通过这些步骤,我们可以确定一种物质的晶体结构,从而进一步深入理解其性质和相互作用。

晶体结构解析的过程1、挑选直径大约为0.1 1.0mm的单晶。
CCD的准直管直径有0.3mm,0.5mm,0.8mm;分别对应得晶体大小是0-0.3mm, 0.3-0.5mm, 0.5-0.8mm.2、选择用铜靶还是钼靶?铜靶要求θmax〉=66度,最大分辨率是0.77埃钼靶要求θmax〉=25度,最大分辨率是0.36埃3、用smart程序收集衍射数据:得到大约一千张倒易空间的衍射图像,300M大小。
其中matrix图像45张,分成三组,每组15张,用以判定晶体能否解析。
4、用saint程序还原衍射数据:得到很多文件,但是只有三个文件是我们需要的:-ls,p4p,raw。
-ls文件中包含有最大的和最小的θ角,有效地精修衍射点数目。
好像不同的机器或者还原程序得到的文件不同,有的是hkl,abs。
5、用shelxtl程序处理上述数据,并画出需要的图形。
5.1 装好shelxtl程序,新建一个project,输入要建立工程的名字,然后打开要解析的p4p或者raw文件。
5.2 用xprep程序确立空间群,建立指令文件这个过程基本上是一直按回车键的过程(除了在要输入化学成分的时候改动一下和在是否建立指令文件的时候输入Y即可),一般不会出错。
如果出错,那就要重新对空间群进行指认(出错可能是出现在下面的精修过程中)。
一般Mean(I/sigma)〉2才可以,越大越好。
得到ins,hkl,pcf三个重要数据文件。
其中ins文件:包含分子式,空间群等信息;hkl文件:包含的是衍射点的强度数据;pcf文件:记录了晶体物理特征,分子式,空间群,衍射数据收集的条件以及使用的相关软件等信息。
5.3 选择要解析的方法:直接法(TREF)还是帕特深法(PATT)?如果晶体中含有重原子如金属原子,那就要用PATT法;如果晶体中没有原子量差异特别大的原子,就用TREF法。
默认的方法是直接法。
5.4 用xs程序解析粗结构得到res文件:包含了ins文件的内容和所有的Q峰信息。

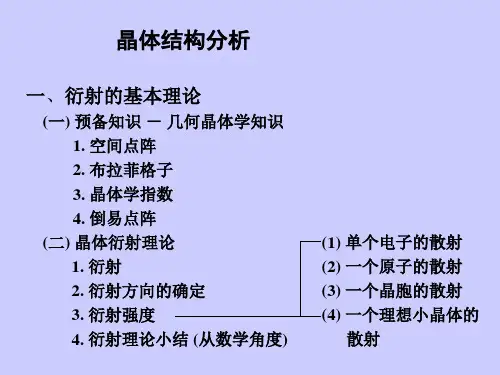

晶体结构分析是研究物质晶体结构的理论和实验方法,属于物理学、化学及材料学等多个领域中的重要分支。
是解决材料科学、物理学和化学等各领域的研究问题的基础和前提,具有十分重要的科学价值和实际意义。
一、晶体结构的基本概念晶体是一种物质的有序形式,具有规律的几何外观。
晶体的形成过程可以去掉杂质并稳定其结构。
一个物质的晶体结构是指这一物质的晶体中所有原子、离子、分子的有序排列和空隙的特殊六面体格局。
晶体的结构是的理解和研究复杂分子间作用的重要途径。
二、晶体结构的分析方法1. X射线衍射法X射线衍射法是分析晶体结构的最重要方法之一。
它是利用X射线通过晶体后产生的衍射图案来研究晶体的方法。
通过X射线的散射和衍射线的特征,可以确定晶体中原子、离子或分子的布局。
这种方法可以得到晶胞的大小、形状和晶体原子的空间位置,适用于金属、无机物和有机物的分析。
2. 中子衍射法中子衍射法也是一种分析晶体结构的重要方法,它是利用中子束通过晶体后产生的衍射图案来研究晶体的方法。
相比X射线衍射,中子衍射的波长更长,穿透力更强,对于轻核元素和氢的散射更敏感,因此可以更容易地分析一些难以通过X 射线分析的样品。
3. 电子衍射法电子衍射法是使用电子束通过晶体后产生的衍射图案来研究晶体的方法。
该技术可用于厚度不超过数百埃的晶体的结构分析。
它的主要限制是,样品必须是真空中,并且必须小于1 mm。
三、的应用可以应用于诸如材料学、生物学、地球化学、制药、高分子化学、金属材料学等多种领域。
它可以用来解决许多问题,例如:1. 建立物质中原子、离子、分子的距离和角度,帮助分子合成和元素结构分析。
2. 获得材料确定结构、确定与路线,来控制各种材料的性能和性质。
3. 研究藏石、宝石和人造宝石的结构和特性,以及开发金刚石和蓝宝石等人工制品的应用。
4. 帮助构建大分子间作用的理论模型,进而实现新型高分子材料的合成和设计。
5. 揭示结晶生长的机制,帮助改进化学反应器和生物发酵技术。


目录一、结构解析的过程(一)空间群的确定(二)解初结构(三)结构精修1、结构精修2、检验精修完毕的参考标准3、精修时INS文件中的指令和意义4、CIF文件5、用WinGX生成键长键角表二、画图1、XP中的指令2、操作实例三、H键分析1、策略2、步骤3、实例四、芳香环间的相互作用1、作用模型2、判断芳香环间相互作用的步骤3、实例五、CIF格式一、结构解析的过程WinGX程序平台集成了下列主要程序:1、确定空间群 (XPREP)2、解初结构(SHELXS-97、SIR-92、SIR-97、SIR-2002)3、结构精修 (SHELXL)(一)空间群的确定将衍射实验得到.hkl和.p4p两个文件(名称应一致)拷入一个文件夹中。
打开WinGX, 从标题栏File命令中选择CHANGE PROJECT下的Select New Project, 此时会出现一个对话框,添加上述的.hkl或.p4p文件。
1)标题栏Data命令中选择Xprep, 出现一个新的对话框,输入.hkl的文件名。
2)出现Select option命令,默认[4]。
3)出现Mean(I/sigma)代表平均信/噪比(该数值要求>7,12~20之间比较好)和格子类型。
默认格子类型即可。
4)选择H:Search for higher Metric Symmetry,寻找更高的对称性。
5)程序显示各种可能的晶系和格子类型。
根据R int值大小确定晶系和格子类型。
6)选择S:Determine or input space group,测定或输入空间群。
7)选择S:Determine space group,测定空间群。
8)程序提供可能的晶系选择,认同程序的选择即可。
9)程序列出所选晶系下的可能晶格类型。
10)选定晶格类型后,程序将列出各种可能的空间群。
11)选定空间群后,程序提示下一选项:Define unit-cell CONTENTS (给出化合物的组成)。
晶体结构解析基本步骤
1.实验数据收集:首先,需要通过实验方法收集晶体的衍射数据。
目
前常用的晶体衍射方法主要有X射线衍射和电子衍射。
实验数据收集可以
获得晶体表面上的几何信息以及衍射角度。
2.数据处理:在收集到的实验数据中,通常需要进行一些运算和处理,以获得有用的衍射信息。
这些数据处理方法包括背景减除、数据标定和归
一化等。
3.数据解析:数据解析是指根据衍射数据推导晶体结构的过程。
解析
晶体结构的方法主要有直接方法、间接方法和混合方法。
直接方法是指通
过衍射数据直接得到晶体的电子密度分布。
间接方法则是通过比较实验衍
射数据和模拟衍射数据,对晶体结构进行推导。
混合方法则是结合了直接
方法和间接方法的优点。
4.模型建立:在获得了晶体结构的初步解析结果后,需要进一步建立
晶体结构的模型。
模型的建立可以利用计算化学方法进行优化,以获得最
稳定的晶体结构。
5.结构验证:结构解析的最后一步是验证得到的晶体结构是否准确。
结构验证可以通过与其他实验数据的比较、理论计算的对比以及晶体学的
规则判断来完成。
除了上述的基本步骤外,晶体结构解析还需要依赖于大量的晶体学理
论知识和计算化学方法。
在解析过程中,还需要考虑晶体的对称性和特殊
性质,以及可能存在的结构缺陷和杂质等问题。
总之,晶体结构解析是一项复杂而精密的研究工作,需要运用物理、
化学、数学等多学科的知识和方法。
通过对晶体结构的解析,可以深入理
解固体物质的性质和行为,从而为材料科学和工程技术提供重要的参考和应用基础。
单晶结构解析技巧1. 通常,H原子的处理方法作者要给出:(1)一般通过理论加H,其温度因子为固定值,可通过INS等文件查看(2) 水分子上H原子可通过Fourier syntheses得到(3)检查理论加上的H原子是否正确,主要看H原子的方向。
若不正确则删去再通过Fourier syntheses合成得到(4) 检查H原子的键长、键角、温度因子等参数是否正常。
通过检查分子间或分子内的H键是否合理最易看出H键的合理性(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)(6) 各位来说说方法与心得?2.胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio222_ALERT_3_B Large Non-Solvent H Ueq(max)/Ueq(min) ... 4.97 Ratio342_ALERT_3_B Low Bond Precision on C-C bonds (x 1000) Ang (49)B 级提示当然得重视了。
建议你先把H撤消,精修到C的热椭球不太变形和键长趋正常。
如做不到就要看空间群?衍射点变量比太小?以至追查到原始数据的录取参数和处理等。
这些粗略意见仅供参考,如何?3.在XP中画图时,只有一部分,想长出另外的对称部分。
我是envi完了,然后sgen长出来的,可是和symm显示的对称信息不一样。
比如:我根据envi的结果用sgen O1 4555得到的是O1A而不是O1D,这跟文献中标注的不一样啊,怎么统一呢?很困扰,忘达人指教。
xp里是按顺序编号的,第一个sgen出的的统一为A,依次标号。
你如果想一开始就统一D的话,重新name一下4.高氯酸根怎么精修呀?我用的SHETXL6.1版的,最好告诉我怎么用其中的XSHELL来做,我觉得他好用!Method 1DFIXDfix 1.42 0.02 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Dfix 1.42 0.02 O1 O2 O1 O3 O1 O4 O2 O3O2 O4O3 O4Method 2SADISadi 0.01 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Sadi 0.01 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O45. 晶体的无序是怎么造成的呀,是晶体培养的问题吗?如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5大于0.5没什么的,解完后都在1以下就可以了。
单晶结构解析过程
单晶结构解析过程是指通过实验和数据分析来确定晶体中原子的位置、晶格参数和晶体结构的方法。
下面是单晶结构解析的常见步骤:
1. 晶体生长:首先需要获得足够大的单晶样品。
这可以通过各种方法实现,如溶液法、气相法或熔融法。
2. 数据收集:使用X射线衍射技术或中子衍射技术,将单晶样品放置在仪器中,并记录衍射图案。
这些衍射数据包含了不同角度的散射强度和相位信息。
3. 数据处理:对收集到的衍射数据进行处理和分析。
其中一个关键步骤是解析Laue图或斑图,确定晶体的晶系和对称性。
4. 相位问题:由于晶体中的散射信息只包含幅度而没有相位,所以需要采用一些方法来解决相位问题。
常见的方法包括多晶片、重组法、直接法和Patterson法等。
5. 结构求解:根据已解决的相位问题,借助计算机软件或手动计算,进行晶体结构求解。
这个过程包括模型建立、参数优化和误差分析等。
6. 结构修正:对求解得到的初始结构进行修正和调整。
这可能涉及到原子位置的微调、氢原子的添加、电荷密度修正等。
7. 结果验证:最后,通过一系列实验数据和计算方法来验证所得到的晶体结构。
这些包括衍射数据与计算模型之间的比较,键长和键角的合理性,以及物理化学性质的一致性等。
晶体结构解析基本步骤Steps to Crystallographic Solution(基于SHELXL97结构解析程序的SHELXTL软件,尚需WINGX和DIAMOND程序配合)注意:每一个晶体数据必须在数据所在的目录(E:\STRUCT)下建立一子目录(如E:\STRUCT\AAA),并将最初的数据备份一份于AAA目录下的子目录ORIG,形成如右图所示的树形结构。
一. 准备1. 对IP收录的数据, 检查是否有inf、dat和f2(设为sss.f2, 并更名为sss.hkl)文件; 对CCD 收录的数据, 检查是否有同名的p4p和hkl(设为sss.hkl)文件2. 对IP收录的数据, 用EDIT或记事本打开dat或inf文件, 并于记录本上记录下相关数据(下面所说的记录均指记录于记录本上):⊕从% crystal data项中,记下晶胞参数及标准偏差(cell);晶体大小(crystal size);颜色(crystal color);形状(crystal habit);测量温度(experiment temperature);⊕从total reflections项中,记下总点数;从R merge项中,记下Rint=?.???? % (IP收录者常将衍射数据转化为独立衍射点后传给我们);⊕从unique reflections项中,记下独立点数对CCD收录的数据, 用EDIT或记事本打开P4P文件, 并于记录下相关数据:⊕从CELL和CELLSD项中,记下晶胞参数及标准偏差;⊕从CCOLOR项中,记下晶体颜色; 总点数;从CSIZE项中,记下晶体大小;⊕从BRA V AIS和SYMM项中,记下BRA V AIS点阵型式和LAUE群3. 双击桌面的SHELXTL图标(打开程序), 呈4. 单击Project New, 先在“查找范围”选择数据所在的文件夹(如E:\STRUCT\AAA), 并选择衍射点数据文件(如sss.hkl), 最后在“project name”中给一个易于记忆和区分的任务名称(如050925-znbpy). 下次要处理同一结构时, 则只需Project Open, 在任务项中选择050925-znbpy便可5. 单击XPREP , 屏幕将显示DOS式的选择菜单:⊕对IP收录的数据, 输入晶胞参数后回车(下记为<cr>) (建议在一行内将6个参数输入, 核对后<cr>)⊕在一系列运行中, 注意屏幕内容(晶胞取向、格子型式、消光规律等), 一般的操作动作是按<cr>。
晶体结构知识汇总及解题方法技巧一、晶胞中质点的占有率在一个晶胞结构中出现的多个原子,并不是只为这一个晶胞所独立,而是为多个晶胞共用,所以每一个晶胞只能按比例分摊。
分摊的根本原则:晶胞任意位置上的原子如果是被n 个晶胞所共有,则每个晶胞只能分得这个原子的n1。
立方晶胞,顶点上的粒子占棱上的粒子占面上的粒子占体心的粒子占二、常见晶胞分析1. NaCl 型⑴每个晶胞占有 个Na+, 个Cl-,即 个NaCl 粒子⑵每个Na +周围有 个Cl -,每个Cl -周围有 个Na +,与一个Na +距离最近且相等的Cl -围成的空间构型为 。
每个Na +周围与其最近且距离相等的Na +有 个。
⑶0.585g NaCl 晶体(0.01mol )含有 个晶胞。
⑷若已知Na+与Cl-的最短距离为a cm ,则NaCl 晶体的密度为 。
2. CsCl 型⑴在CsCl 晶体中,每个Cs+周围与之最接近的且距离相等的Cs+有 个,每个Cs+周围与之最接近的且距离相等的Cl-有 个。
⑵每个晶胞占有 个CsCl 粒子。
3. 干冰型在干冰晶体中,每个CO2分子周围与之最接近的且距离相等的CO2分子有个。
每个晶胞中含有个CO2分子。
4.金刚石型金刚石的网状结构中,,每个碳原子与其他4个碳原子等距离紧邻,含有由共价键形成的碳原子环,其中最小的环上有6个碳原子,每个碳原子上的任意两个C—C键的夹角都是109°28′,其中C原子个数:C—C个数= 。
5.石英晶体在二氧化硅晶体中,一个硅原子和4个氧原子形成4个共价键,1个氧原子和2个硅原子形成2个共价键,故Si原子与O原子数目之比为。
实际上,该晶体是由硅原子和氧原子按1:2的比例组成的立体网状晶体,没有单个分子存在。
在晶体中最小的环为十二元环,每个环占有6个Si原子和6个O原子。
6.石墨晶体结构石墨晶体是一种混合型晶体,层内存在共价键,层间以范德华力结合,兼具有原子晶体、分子晶体的特征和特性。
晶体结构解析步骤Steps to Crystallographic Solution(基于SHELXL97结构解析程序和DOS版SHELXTL画图软件。
在DOS下操作)注意:1. 每一个晶体数据必须在D:/STRUCT下建立一子目录(如D:\STRUCT\AAA),并将最初的数据备份一份于AAA目录下的子目录ORG;2. 此处用了STRUCT.BA T批文件,它存在于C:\根目录下,内有path= c:\nix; c:\exe; d:\ struct; c:\windows\system32 (struct为工作目录,exe为SHELXL97程序,nix为SHELXTL画图)3. 在了解DOS下操作之后,可在WIN的WINGX界面下进行结构解析工作,画图可用XP 或DIAMOND软件进行。
一. 准备1. 检查是否有inf、dat和f2(设为sss.f2)文件2. 用EDIT或记事本打开dat或inf文件, 并于记录本上记录下相关数据(下面所说的记录均指记录于记录本上):⊕从% crystal data项中,记下晶胞参数及标准偏差(cell);晶体大小(crystal size);颜色(crystal color);形状(crystal habit);测量温度(experiment temperature);⊕从R merge项中,记下Rint=?.???? %;⊕从total reflections项中,记下总点数;⊕从unique reflections项中,记下独立点数3. 双击桌面的DOS图标(或Win2000与WinNT的“命令提示符”)4. 键入STRUCT(属于命令,大小写均可。
下同)5. 进入欲处理的数据所在的文件夹(上面的1~2工作也可在这之后进行)6. 键入XPREP sss.f2 (屏幕显示DOS的选择菜单)7. 选择[4],回车(下记为)8. 输入晶胞参数(建议在一行内将6个参数输入,核对后)9. 一系列运行(对应的操作动作均为按)之后,输入分子式(如, Cu2SO4N2C4H12。
此分子式仅为估计之用。
注意:反应中所有元素都应尽可能出现,以避免后续处理的麻烦)10. 退出XPREP运行之前,机器要求输入文件名,此时一定要输入文件名,且不与初始的文件名同名。
另外,不要输入扩展名。
如可输入aaa11. 检查是否产生有PRP、PAR和INS文件(PRP文件内有机器对空间群确定的简要说明)12. 更名:REN aaa.f2 aaa.hkl13. 用EDIT或记事本打开aaa.ins文件,在第二~三行中,用实际的数据更改晶胞参数及其偏差(注意:当取向改变了,晶胞参数也应随之对应),波长用实际波长。
二.解结构14. 键入SHELXS aaa或XS aaa,(INS文件中, TREF为直接法,PATT为Pattersion法)15. XP,(进入XP程序)(可能产生计算内址冲突问题,注意选择处理)16. READ or REAP aaa (aaa.res 为缺省值,若其它文件应是文件名.扩展名,如aaa.ins)17. FMOL, (不要H原子时,为FMOL LESS $H,或FMOL后,KILL $H, ) (读取各参数,屏幕上显示各原子的键合情况)18. MPLN/N, (机器认为最好取向)19. PROJ, (随意转动,直至你认为最理想取向)20. PICK,(认为合理的位置投相应原子,如C原子键入C8,注意序号不能重复;不合理的用剔除,暂时不确定用空格键放弃,完成或不再投原子时键入"/")21. SORT …….. (排序) 如,SORT $Cu $N $C $H22. FILE aaa, (保存文件)23. EXIT,或QUIT,(退出XP程序)24. 打开aaa.ins,删去原子序号之前的HKLF 4这一行以及机器自动产生的REM行(借助WINGX界面不会产生这一问题)25. 键入SHELXL aaa (此时的目的是用Fourier峰和差值Fourier峰找其它原子)26. 用EDIT或记事本打开aaa.LST文件(查看Fourier峰的大小,记住峰值大于5e/Å3以上的峰如Q1~Q8;如果前一次处理中有误,可能提示一些信息于文件中,请注意处理,去误存真)27. 进行15~23步,投Qn(Fourier峰较大者)28. 重复25~27步骤,直到解出完整结构模型(过程中可打开LST文件查看进行情况)三.结构修正29. 用EDIT打开aaa.ins,并完成:⊕删去原子序号之前的HKLF 4这一行(修正时,不允许在原子序号前有HKLF 4;但HKLF n可放在END的前一行), 以及机器自动产生的REM行;⊕在UNIT行后加入TEMP ?? (单位已设为°C)一行;⊕加入SIZE (晶体的三维尺寸???? ???? ????,单位已设为mm)一行;⊕在MERG 2前加REM;在OMIT 4前加REM30. 键入SHELXL aaa31. 用EDIT打开aaa.res,并将END后的WGHT行移到FV AR行之后,另存为同名的INS 文件作为输入的指令文件32. 重复30~31步骤,完成同性修正。
每一次修正后,均可打开LST文件查看运行情况。
认为合理时,COPY aaa.res zzz.res (另存文件,以备用)。
必要时,记下同性(此时)的R1和wR2因子(不要求完全收敛)33. 用EDIT打开aaa.res,并将END后的WGHT行移到FVAR行之后,并在原子序号之前加入单独的ANIS n一行(ANIS为异性修正,n为原子个数,可根据情况设定),存为同名的INS文件(履盖了原INS文件)34. SHELXL aaa35. 打开LST或RES文件,不合理时,修正INS文件ANIS n的n, 重复进行32~33步,直到合理后进行第36步36. 进入XP程序,并加H。
对C、N可理论加H,指令为HADD;对其它原子,则需采用Fourier加H,即第16步为REAP aaa ,第20步时投H原子37. FILE aaa, (保存文件)38. EXIT,(退出XP程序)39. 用EDIT打开aaa.ins,并完成:⊕更改BOND 0.5为BOND $H⊕将各个H原子移到相应原子之后,在H原子前加AFIX n3一行,H原子之后加AFIX 0一行(有关n的规定,请查看SHELXL说明书);⊕删去原子序号之前的HKLF 4这一行(修正时,不允许在原子序号前有HKLF 4),以及机器自动产生的REM行;⊕如果非N、C原子仍不能找出H原子,可提高PLAN后的数值(残峰数目多一点)40. 重复29~30和34~38步,继续修正,以致H尽可能全部找出(如果实在找不出,应在记录本上说明具体情况)41. 重复29~30步,直到R1收敛,Shift/error最小(0.00?),残余峰小(<1e/Å3)。
此时,结构精修工作完成。
强烈建议将这时的文件另存一备份(可履盖同性备份文件),COPY aaa.res zzz.res四.氢键查找氢键查找有多种方法,这里只是操作最简单的机器自动产生方法42. 用EDIT打开aaa.res,END后的WGHT行移到FV AR行之后,并在UNIT行后加入HTAB 一行,存为同名的INS文件(履盖了原INS文件)43. SHELXL aaa44. 打开LST文件,记录下H键情况。
并将H键情况拷贝到RES文件中的原子序号之前、BOND之后,并将它们编成HTAB O2 N2_$1形式($1为对称性代码,用EQIV在HTAB之前定义),存编辑后的RES文件为INS文件(履盖了原INS文件)45. SHELXL aaa46. 用EDIT打开LST文件,核查用HTAB a b产生的H键是否全了和准确(和机器HTAB 产生的H键对比)(用HTAB a b产生的H键和用机器HTAB产生的H键的差别在于前者可直接写入CIF文件中并在以后的处理中自动产生H键表,而后者不能;同时,有时机器HTAB 产生的H键不合理,如一个给体同时与三个或以上受体产生H键,这必须依据H键的键长和键角用人工HTAB a b方式挑出正确的)47. 记录下H键情况,以备画图之用五.产生晶体学表48. 用EDIT打开含H键命令的aaa.res,并完成:⊕END后的WGHT行移到FV AR行之后;⊕去掉LIST 行;⊕在UNIT行之后加ACTA一行(以产生晶体学文件CIF和FCF)⊕在ACTA行之后加TEMP(测量温度)(21°C, 为TEMP 21)、SIZE各一行⊕可在CONF前加REM⊕另存为同名的INS文件(履盖了原INS文件)49. SHELXL aaa (运行之后将产生CIF和FCF两个文件)50. 用EDIT打开CIF文件,输入晶系、空间群、颜色、形状、总衍射点数和Rint等参数51. CIFTAB aaa (键入命令之前,可先关闭打印机,以免产生错误动作)T…注意:实验室统一规定,晶体学数据文本文件统一存为aaa.TXT;衍射点文件统一为aaa.SFT52. 用EDIT打开aaa.res,在ACTA命令前加REM,以免产生误操作,增加CIF文件48步的编辑工作。
至此,结构解析工作基本完毕(结构解析还包括最小二乘平面的计算MPLA、画图等工作)六.画结构图53. 进行15~19步骤。
其中,17步为READ ZZZ.RES(RES可省。
ZZZ.RES为H键产生前的文件)54. 画ORTEP图:JOIN 5 $H;LABL 1 550;TELP 0 -5055. 画堆积(PACKING)图:MA TR n(n = 1,2或3);PBOX;PACK(其中有许多操作,最后记得选sgen/mol后退出); LABL 1 330; TELP CELL56. 画特殊图*********************************实验室对原子的颜色和线条(ATYP)统一规定为:ATYP n $atom: n=1 for C; 2 for O; 3 for N; 4 for V, Mo or W;n=5 for Cu, Ni, etc. 8 for Cl or X; 3 for tetrahedron 7 for octahedron.注意:画图的指令很多,可通过HELP获得帮助。
每一图都应输入一文件名,文件名应一目了然。
如:BALL为球棒图;ELL50(.PLT)为ORTEP图;PKA为沿a轴的投影图;PKB为沿b轴的投影图;PKC为沿c轴的投影图;HA为沿a轴的H氢键图;1D为一维链图等。
如果是随意的,应在记录本上记下对应文件名的意义。