利用SeqMan进行序列拼接
- 格式:pptx
- 大小:1.38 MB
- 文档页数:34

利用SeqMan进行序列拼接利用SeqMan进行序列拼接Step1:打开Seqman软件Step2:加入你要拼接的序列点击Add sequences查找并选中要拼接的序列(可按住control键进行多选)点击Add按钮填加选择的序列填加完后点击done注:最好用测序的图谱尽量不要直接用测序得到的序列Step3:去除末端序列主要是去除序列末端测序质量差或是载体序列有两种方法可以用来去除这类末端序列其一:利用Seqman自带的去除工具自动去除(利用Trim ends 按钮进行)其二:手工去除个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示手工去除侧翼序列双击要去除侧翼序列的目标序列将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接此步请将测序不准确或认为是载体的序列用这种方法去除。
测序准确的峰形图峰形规则,一般在序列的中部,如下图所示测序不准确的峰形图峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示Step4:进行序列拼接点击Assemble按钮在新出现窗口处点击拼接好的contig1在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图Step5:查找拼接错误find conflict 点击菜单Edit点击Find Previous或Find Next查找接接中出现的错误还可以通过Seqman左下角的快捷按钮查找错误的拼接查找错误的拼接错误的拼接的类型类型1:两条序列的测序结果不一致并明显一条测序质量好而另一条质量差处理:直接将该处修改为正确的碱基类型2:两条序列的测序结果不一致并两条测序质量都比较差处理:重新测序或用新的合适引物重新测定类型3:两条序列的测序结果不一致并明显两条测序质量都好处理:测序过程出现问题,重新测定Step6:导出拼接的序列可选择合适的格式,导出拼接好的序列通过以上几步我们就能很快将几个测序片段进行拼接,大家可以拿着自己的序列试试!当然如果两个测序片段的拼接片段太短可能利用默认的参数不能完成拼接,大家可以试着修改一下拼接参数试试!如降低Match size及Minimum Match Percentage的值!修改参数命令。

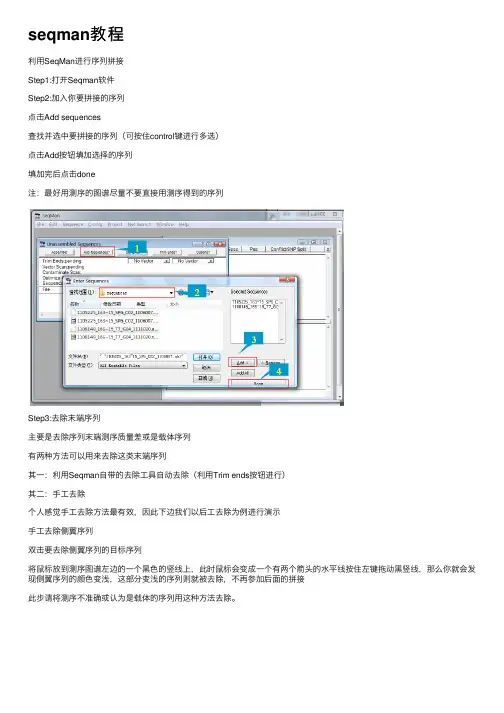
seqman教程利⽤SeqMan进⾏序列拼接Step1:打开Seqman软件Step2:加⼊你要拼接的序列点击Add sequences查找并选中要拼接的序列(可按住control键进⾏多选)点击Add按钮填加选择的序列填加完后点击done注:最好⽤测序的图谱尽量不要直接⽤测序得到的序列Step3:去除末端序列主要是去除序列末端测序质量差或是载体序列有两种⽅法可以⽤来去除这类末端序列其⼀:利⽤Seqman⾃带的去除⼯具⾃动去除(利⽤Trim ends按钮进⾏)其⼆:⼿⼯去除个⼈感觉⼿⼯去除⽅法最有效,因此下边我们以后⼯去除为例进⾏演⽰⼿⼯去除侧翼序列双击要去除侧翼序列的⽬标序列将⿏标放到测序图谱左边的⼀个⿊⾊的竖线上,此时⿏标会变成⼀个有两个箭头的⽔平线按住左键拖动⿊竖线,那么你就会发现侧翼序列的颜⾊变浅,这部分变浅的序列则就被去除,不再参加后⾯的拼接此步请将测序不准确或认为是载体的序列⽤这种⽅法去除。
测序准确的峰形图峰形规则,⼀般在序列的中部,如下图所⽰测序不准确的峰形图峰形较乱,很难判断是哪个碱基,⼀般位于序列两端,如下图所⽰Step4:进⾏序列拼接点击Assemble按钮在新出现窗⼝处点击拼接好的contig1在出现的Alignment of contig1 窗⼝中点击左三⾓显⽰序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图Step5:查找拼接错误find conflict点击菜单Edit点击Find Previous或Find Next查找接接中出现的错误还可以通过Seqman左下⾓的快捷按钮查找错误的拼接查找错误的拼接错误的拼接的类型类型1:两条序列的测序结果不⼀致并明显⼀条测序质量好⽽另⼀条质量差处理:直接将该处修改为正确的碱基类型2:两条序列的测序结果不⼀致并两条测序质量都⽐较差处理:重新测序或⽤新的合适引物重新测定类型3:两条序列的测序结果不⼀致并明显两条测序质量都好处理:测序过程出现问题,重新测定Step6:导出拼接的序列可选择合适的格式,导出拼接好的序列通过以上⼏步我们就能很快将⼏个测序⽚段进⾏拼接,⼤家可以拿着⾃⼰的序列试试!当然如果两个测序⽚段的拼接⽚段太短可能利⽤默认的参数不能完成拼接,⼤家可以试着修改⼀下拼接参数试试!如降低Match size及Minimum Match Percentage的值!修改参数命令。

利用SeqMan进行序列拼接Step1:打开Seqman软件Step2:加入你要拼接的序列点击Add sequences查找并选中要拼接的序列(可按住control键进行多选)点击Add按钮填加选择的序列填加完后点击done注:最好用测序的图谱尽量不要直接用测序得到的序列Step3:去除末端序列主要是去除序列末端测序质量差或是载体序列有两种方法可以用来去除这类末端序列其一:利用Seqman自带的去除工具自动去除(利用Trim ends按钮进行)其二:手工去除个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示手工去除侧翼序列双击要去除侧翼序列的目标序列将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接此步请将测序不准确或认为是载体的序列用这种方法去除。
测序准确的峰形图峰形规则,一般在序列的中部,如下图所示测序不准确的峰形图峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示Step4:进行序列拼接点击Assemble按钮在新出现窗口处点击拼接好的contig1在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图Step5:查找拼接错误find conflict点击菜单Edit点击Find Previous或Find Next查找接接中出现的错误还可以通过Seqman左下角的快捷按钮查找错误的拼接查找错误的拼接错误的拼接的类型类型1:两条序列的测序结果不一致并明显一条测序质量好而另一条质量差处理:直接将该处修改为正确的碱基类型2:两条序列的测序结果不一致并两条测序质量都比较差处理:重新测序或用新的合适引物重新测定类型3:两条序列的测序结果不一致并明显两条测序质量都好处理:测序过程出现问题,重新测定Step6:导出拼接的序列可选择合适的格式,导出拼接好的序列通过以上几步我们就能很快将几个测序片段进行拼接,大家可以拿着自己的序列试试!当然如果两个测序片段的拼接片段太短可能利用默认的参数不能完成拼接,大家可以试着修改一下拼接参数试试!如降低Match size及Minimum Match Percentage的值!修改参数命令。


测序后的序列为两种形式:abi,seq
abi:波峰图seq:atcg序列
seqman→file→new→skip→add sequences→选择一对引物的.seq和.abi 格式的文件双击(或者选中文件→add)→done→assemble→双击右侧的conting→看峰的好坏进行裁剪→contig→save consensus→single file→命名保存为.fas格式→file→close
mega比对
用mega打开所需比对的文件,如果要添加序列可以选中最下面一个基因序列的一个碱基,右键copy。
选中一个碱基→W→align DNA→OK→OK
→alignment→align by clustalw→OK→OK
将比对完的数据另存为mega格式
用mega打开该文件
点击TA
C:保守位点V:变异位点Pi:简约信息位点S:单个位点0 :0倍退化位点 2 :2倍退化位点 4 :4倍退化位点Statistics→nucleotide composition 核苷酸组成
Distance→compute pairmise distance…→OK→compute两两遗传距离。
利用SeqMan进行序列拼接Step1:打开Seqman软件Step2:加入你要拼接的序列点击Add sequences查找并选中要拼接的序列(可按住control键进行多选)点击Add按钮填加选择的序列填加完后点击done注:最好用测序的图谱尽量不要直接用测序得到的序列Step3:去除末端序列主要是去除序列末端测序质量差或是载体序列有两种方法可以用来去除这类末端序列其一:利用Seqman自带的去除工具自动去除(利用Trim ends按钮进行)其二:手工去除个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示手工去除侧翼序列双击要去除侧翼序列的目标序列将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接此步请将测序不准确或认为是载体的序列用这种方法去除。
测序准确的峰形图峰形规则,一般在序列的中部,如下图所示测序不准确的峰形图峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示Step4:进行序列拼接点击Assemble按钮在新出现窗口处点击拼接好的contig1在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图Step5:查找拼接错误find conflict 点击菜单Edit点击Find Previous或Find Next查找接接中出现的错误还可以通过Seqman左下角的快捷按钮查找错误的拼接查找错误的拼接错误的拼接的类型类型1:两条序列的测序结果不一致并明显一条测序质量好而另一条质量差处理:直接将该处修改为正确的碱基类型2:两条序列的测序结果不一致并两条测序质量都比较差处理:重新测序或用新的合适引物重新测定类型3:两条序列的测序结果不一致并明显两条测序质量都好处理:测序过程出现问题,重新测定Step6:导出拼接的序列可选择合适的格式,导出拼接好的序列通过以上几步我们就能很快将几个测序片段进行拼接,大家可以拿着自己的序列试试!当然如果两个测序片段的拼接片段太短可能利用默认的参数不能完成拼接,大家可以试着修改一下拼接参数试试!如降低Match size及Minimum Match Percentage的值!修改参数命令。

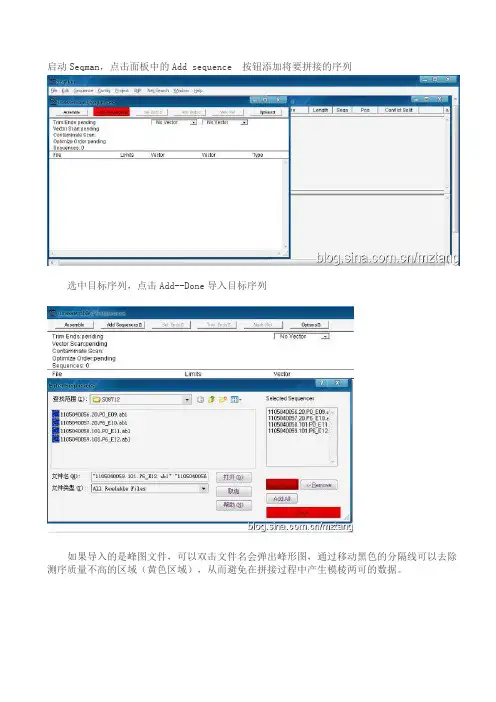
启动Seqman,点击面板中的Add sequence 按钮添加将要拼接的序列
选中目标序列,点击Add--Done导入目标序列
如果导入的是峰图文件,可以双击文件名会弹出峰形图,通过移动黑色的分隔线可以去除测序质量不高的区域(黄色区域),从而避免在拼接过程中产生模棱两可的数据。
数据修正后点击Assemble即拼接,运行结束后会弹出一个新对话框,如果能拼接上,则在Contig一栏有显示。
双击Contig可以看到拼接好的完整序列,以及两个峰图的重叠区域,如果两个峰图的重叠区域完全匹配,则表明拼接的结果可靠;如果两个峰图间有不同碱基,则需要比对两个峰图,选择峰图清晰的作为最终结果。
点击文件前面的三角形可以直接查看序列的峰形图
点击菜单栏的Contig--Save Consense--Single File 可以保存拼接好的序列。

DNAstar软件的使用(三)Seqman 拼接序列
启动Seqman,点击面板中的Add sequence按钮添加将要拼接的序列
选中目标序列,点击Add--Done导入目标序列
如果导入的是峰图文件,可以双击文件名会弹出峰形图,通过移动黑色的分隔线可以去除测序质量不高的区域(黄色区域),从而避免在拼接过程中产生模棱两可的数据。
数据修正后点击Assemble即拼接,运行结束后会弹出一个新对话框,如果能拼接上,则在Contig一栏有显示。
双击Contig可以看到拼接好的完整序列,以及两个峰图的重叠区域,如果两个峰图的重叠区域完全匹配,则表明拼接的结果可靠;如果两个峰图间有不同碱基,则需要比对两个峰图,选择峰图清晰的作为最终结果。
点击文件前面的三角形可以直接查看序列的峰形图
点击菜单栏的Contig--Save Consense--Single File 可以保存拼接好的序列。
启动Seqman,点击面板中的Add sequence 按钮添加将要拼接的序列
选中目标序列,点击Add--Done导入目标序列
如果导入的是峰图文件,可以双击文件名会弹出峰形图,通过移动黑色的分隔线可以去除测序质量不高的区域(黄色区域),从而避免在拼接过程中产生模棱两可的数据。
数据修正后点击Assemble即拼接,运行结束后会弹出一个新对话框,如果能拼接上,则在Contig一栏有显示。
双击Contig可以看到拼接好的完整序列,以及两个峰图的重叠区域,如果两个峰图的重叠区域完全匹配,则表明拼接的结果可靠;如果两个峰图间有不同碱基,则需要比对两个峰图,选择峰图清晰的作为最终结果。
点击文件前面的三角形可以直接查看序列的峰形图
点击菜单栏的Contig--Save Consense--Single File 可以保存拼接好的序列。