DNA测序结果拼接方法
- 格式:ppt
- 大小:4.51 MB
- 文档页数:6

测序结果说明1.测序完成后,我们用对每个样品提供一份测序报告,其中包括:测出的序列彩色峰图(请用Chromas软件打开)序列文件拼接后结果(需要测通的样品)2.在进行DNA测序时,紧接引物的10~30碱基有时不一定能完全读清楚。
3.正常情况下,3730测序仪保证800bp的有效长度,但是有时由于DNA结构上的原因,有时会出现反应中断无法进行的情况。
如:G/C rich;G/C Cluster;Poly A/T/C/G的连续结构等。
此外,另一种情况为反应中途出现的套峰现象,此种情况一般为DNA结构中的重复序列,造成测序用引物和模板之间有两个以上结合位点。
以上情况是由于DNA结构原因造成了无法正常进行DNA测序,对这种情况,我们会根据具体测序结果进行相应收费。
出现以上情况后,我们提倡从另一端进行测序,或者用高级试剂盒进行测序。
4.备注说明一般正常的菌液和质粒自收样日至发送报告日周期为二--三天,PCR产物(纯化及未纯化)为三--四天。
对于三个工作日内无法得到满意测序结果的,我们会用E-mail或电话与客户或代理商联系。
并不是所有的样品都能得到满意的测序结果,对于测序结果不好的,我们会尽力找到可能的原因,以建议进一步如何操作。
测序常见问题解答Q1.为什么提供新鲜的菌液?如何提供新鲜的菌液?A1.首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度。
如果您提供新鲜菌液,用封口膜封口以免泄露;也可以将培养好的4~5ml菌液离心沉淀下来,倒去上清液以方便邮寄。
同时邮寄时最好用盒子以免邮寄过程中造成离心管挤压破裂。
Q2.DNA测序样品用什么溶液溶解比较好?A2.溶解DNA测序样品时,用灭菌蒸馏水溶解最好。
DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。
如果DNA用缓冲液溶解后,在进行测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。

染色体水平组装基因组染色体水平组装基因组是一种重要的生物学技术,它可以帮助我们更好地理解基因组的结构和功能。
本文将介绍染色体水平组装基因组的原理、方法和应用,并探讨其在生物学研究和医学领域的潜在应用。
染色体水平组装基因组是指通过将测序读段按照染色体上的位置进行组装,重建出完整的染色体序列。
相比于传统的基因组组装方法,染色体水平组装基因组能够提供更长的连续序列,有助于揭示基因组的结构和功能。
染色体水平组装基因组的原理是利用测序技术对DNA分子进行测序,并根据测序结果将读段按照染色体上的位置进行组装。
首先,需要将DNA分子进行打断,并利用测序技术对其进行测序。
然后,根据测序结果将读段按照染色体上的位置进行排序和组装。
最后,通过对组装结果进行验证和校正,得到完整的染色体序列。
染色体水平组装基因组的方法主要包括两个步骤:测序和组装。
测序步骤可以采用多种测序技术,如Sanger测序、Illumina测序和PacBio测序等。
不同的测序技术具有不同的优缺点,可以根据研究的需求选择合适的测序技术。
组装步骤则是将测序读段按照染色体上的位置进行排序和组装,常用的组装算法包括Overlap-Layout-Consensus(OLC)算法和De Bruijn图算法等。
染色体水平组装基因组在生物学研究中具有广泛的应用。
首先,它可以帮助我们理解基因组的结构和功能。
通过组装染色体序列,我们可以了解基因的分布和排列方式,揭示基因组的整体结构和组织方式。
其次,染色体水平组装基因组可以帮助我们研究基因组的进化和变异。
通过比较不同物种的染色体序列,我们可以揭示物种间的遗传差异和进化关系。
此外,染色体水平组装基因组还可以应用于基因组编辑和合成生物学等领域,为基因工程和合成生物学的研究提供重要的工具和方法。
在医学领域,染色体水平组装基因组也具有重要的应用价值。
首先,它可以帮助我们研究人类基因组的结构和功能。
通过组装人类染色体序列,我们可以了解人类基因的分布和排列方式,揭示人类基因组的整体结构和组织方式。

简述一、二、三代测序技术
一代测序技术
一代测序技术是一种拼接式测序技术,它可以将DNA片段进行拼接,从而得到DNA序列。
它是一种基于Sanger方法的技术,通过热板和冷板将DNA片段分别固定在支架上,再使用DNA聚合酶对支架上的DNA片段进行复制,最后通过测序仪来获取DNA序列信息。
一代测序技术已经被广泛应用于基因组学研究中,但是它仍然有很多缺点,比如时间短,费用较高,最大的问题是在测序过程中可能出现错误,这种错误很难被确认。
二代测序技术
二代测序技术是一种新的技术,它不需要DNA片段的拼接,而是使用DNA分子组装的方法来提取DNA序列信息。
该技术使用高通量测序技术,可以一次性同时测序大量的DNA片段,因此大大提高了测序效率,并减少了出错的几率,同时也降低了测序成本。
三代测序技术
三代测序技术是一种后续的测序技术,它能够更加精确地提取DNA序列信息,使用特殊的测序仪可以同时测定全基因组的DNA序列。
该技术采用短片段拼接的方法,可以实现更高精度的DNA序列测序,可以更好地发掘基因组中的变异位点,从而更好地研究遗传病和肿瘤的发生机制。

三代测序拼接算法(原创版)目录1.三代测序拼接算法的背景和意义2.三代测序拼接算法的原理和方法3.三代测序拼接算法的应用案例4.三代测序拼接算法的优缺点和未来发展方向正文三代测序拼接算法是一种在基因组学研究中广泛应用的技术,尤其在处理较长的 DNA 序列拼接上具有重要意义。
本文将从原理、方法、应用案例以及优缺点等方面,详细介绍三代测序拼接算法。
一、三代测序拼接算法的背景和意义随着基因组学研究的深入,研究人员需要对越来越长的 DNA 序列进行拼接。
传统的 Sanger 测序技术由于其局限性,难以应对这种需求。
因此,三代测序拼接算法应运而生,它能够更有效地处理较长的 DNA 序列拼接问题。
二、三代测序拼接算法的原理和方法三代测序拼接算法主要基于 PacBio SMRT 技术,通过构建 SMRT 测序数据和 Hi-C 数据之间的联系,实现长 DNA 序列的拼接。
具体方法包括以下几个步骤:1.构建 SMRT 测序数据和 Hi-C 数据的联系通过比对 SMRT 测序数据和 Hi-C 数据,找到它们之间的匹配区域,从而构建起它们之间的联系。
2.利用联系进行拼接根据构建的联系,将 SMRT 测序数据和 Hi-C 数据进行拼接,得到目标 DNA 序列。
3.拼接结果评估与优化对拼接结果进行评估,通过优化拼接策略和参数,提高拼接的准确性和完整性。
三、三代测序拼接算法的应用案例三代测序拼接算法在多个领域都取得了显著的应用成果,例如:1.人类基因组拼接利用三代测序拼接算法,研究人员成功拼接了人类基因组中的复杂区域,为全面解析人类基因组结构提供了有力支持。
2.动植物基因组拼接三代测序拼接算法在动植物基因组拼接方面也取得了显著成果,为研究动植物基因组结构和功能提供了有力工具。
四、三代测序拼接算法的优缺点和未来发展方向三代测序拼接算法具有以下优缺点:优点:能够有效地处理较长的 DNA 序列拼接问题,提高拼接的准确性和完整性。

1 、为什么开始一段序列的信号很杂乱,几乎难以辨别这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。
2 、为什么在序列的末端容易产生 N 值,峰图较杂由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生 N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。
3 、测序结果怎么找不到我的引物序列如果找不到测序所用的引物序列。
这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。
4 、测序结果怎么看不到我克隆的酶切位点可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。
通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。
5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。
6 、序列图为什么会有背景噪音(杂带)是否会影响测序结果序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。
7 、测序结果为什么与标准序列有差别原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。




单细胞DNA甲基化测序数据处理流程与分析方法1. 内容简述单细胞DNA甲基化测序是一种高分辨率的基因表达和表观遗传学研究方法,它允许研究者检测单个细胞的DNA甲基化状态。
这种技术为理解细胞异质性、基因调控机制以及疾病发展中的表观遗传变化提供了有力工具。
样本制备:首先,从生物体中提取单细胞,然后利用亚硫酸盐转化技术将DNA中的甲基化修饰转换为羟基化修饰,以供后续测序。
文库构建:转化后的DNA被随机打断成小片段,并加上特定的接头序列,以便进行PCR扩增和测序。
测序:构建好的文库被加载到测序芯片上,通过高通量测序技术进行测序。
数据分析:获得的原始数据需要经过一系列清洗、比对、标准化等处理步骤,以获得高质量的甲基化数据集。
甲基化状态分析:识别每个细胞中的甲基化位点,并比较不同细胞之间的甲基化差异。
差异甲基化分析:识别在不同实验条件下(如疾病状态、环境压力等)甲基化模式的差异。
生物信息学分析:使用统计软件和算法对数据进行深度挖掘,发现与特定生物学过程或疾病相关的甲基化模式。
通过对这些数据的综合分析,研究者可以揭示细胞功能的动态变化、基因表达的调控机制以及表观遗传学在疾病发生中的作用。
1.1 单细胞DNA甲基化测序技术简介简称SCDBS)是一种高通量、高分辨率的分析方法,用于研究单个细胞中基因组水平的DNA甲基化状态。
该技术通过测序和分析单细胞中的甲基化位点序列,揭示了基因表达差异、发育过程、疾病发生机制等方面的信息。
随着高通量测序技术的快速发展,SCDBS已经成为生物学研究的重要工具之一。
SCDBS的主要流程包括:样品准备、文库构建、测序、数据处理和分析等步骤。
需要将单细胞样本进行处理,如去除血浆等杂质,保证测序结果的准确性。
通过构建文库来存储待测的DNA片段,通常采用Illumina测序平台进行高通量测序。
对测序数据进行质量控制和过滤,以去除低质量序列和伪迹。
利用生物信息学工具对数据进行处理和分析,包括聚类分析、差异基因表达分析、甲基化模式比较等。

图解DNA测序技术的原理与操作DNA测序技术是一种快速准确地测定DNA序列的方法,它可以应用于基础研究、医学诊断、辅助决策等领域。
DNA测序技术的原理是通过测量DNA碱基之间的化学键来确定其序列。
DNA测序的发展历程DNA测序技术在20世纪中期开始发展。
当时,科学家们利用毒蕈碱和亚硝酸钠等化学试剂将DNA分解成单个核苷酸分子,然后通过电泳将分子按照大小分开。
通过观察电泳结果,科学家们得以确定碱基序列。
20世纪70年代末,Sanger发明了dideoxy法,使得测序技术得到了重大的进展。
dideoxy法以单链DNA为模板,在该模板上合成一系列新的DNA片段。
这些新的DNA片段不同于模板,因为它们以特定的方式截止于dideoxynucleotide。
这些dideoxynucleotide是无法延伸的,因此一旦它们被合成到了新的DNA片段中,该片段合成就截止了。
这种方法可以验证DNA序列,并且可以在大规模基因组测序任务中运用。
现代DNA测序技术现代DNA测序技术有多种,包括Illumina、Ion Torrent、PacBio和Nanopore等。
其中,Illumina是目前最广泛应用的测序技术之一。
Illumina 测序技术Illumina使用悬浮在小珠子上的(End-Bound)DNAs浓缩在测试管中。
在静止的状况下,铺设在黄色胶片上的DNA接头被拉伸,并用荧光标记的ddNTP来扩展从已接头的DNA的3'端到最近发出的碱基。
这种延长作用结合了测序信号和序列测量。
处理过的簇可以被移动到一个新的位置上,这样就可以在具有更高的通量和更高的密度下进行处理。
Illumina紧凑的读片机设计可容纳数以千亿的簇,因此即便少量的DNA也可以实现高通量测序。
DNA测序技术的操作步骤DNA测序的一般操作步骤包括:样品制备、文库构建、测序准备、碱基测序、数据解读和结果分析等。
具体步骤如下:样品制备:将待检测样品提取DNA,并在PCR扩增过程中对DNA进行分离和纯化,以使其达到测序指标。
生物大数据技术中的基因组装方法介绍在生物学研究中,基因组装是一项重要的任务,它的目标是将原始的DNA序列片段拼接起来,以重建完整的基因组序列。
随着技术的进步和生物大数据的爆发式增长,出现了许多新的基因组装方法,这些方法能够更快、更准确地拼接基因组序列。
本文将介绍三种常用的基因组装方法:重叠图法、de Bruijn图法和基于单分子测序的方法。
1. 重叠图法重叠图法是最早也是最传统的基因组装方法之一。
它的原理是通过比较DNA序列片段之间的相似性,找出它们之间的重叠区域,并将这些片段组合起来形成连续的序列。
具体步骤如下:1)寻找重叠区域:将所有的DNA序列片段进行两两比对,找出它们之间的重叠区域。
2)构建重叠图:将找到的重叠区域以节点的形式表示,并连接起来构成一个图,称为重叠图。
3)拼接序列:在重叠图中找到一条路径,它能够覆盖所有的节点,并且使得路径上的序列片段拼接在一起,形成完整的基因组序列。
重叠图法有着简单明了的原理和操作流程,但它在处理大规模数据时效率较低,并且容易受到测序错误和基因组重复序列的干扰。
2. de Bruijn图法de Bruijn图法是一种常用的基于kmer的基因组装方法。
它将DNA序列片段分割成长度为k的kmer,并将kmer作为节点构建一个图,称为de Bruijn图。
具体步骤如下:1)构建kmer集合:将所有的DNA序列片段分割成长度为k的kmer,并将它们作为节点添加到de Bruijn图中。
2)连接节点:根据kmer之间的重叠关系,在de Bruijn图中添加边连接相邻的节点。
3)拼接序列:在de Bruijn图中找到一条欧拉路径,即从一个节点出发,经过所有的节点,每个边只经过一次,最终形成完整的基因组序列。
de Bruijn图法在处理大规模数据时有着较高的效率,并且能够有效解决测序错误和基因组重复序列的问题。
但它在一些特殊情况下,如序列重复率较高或者存在大量的测序错误时,可能会出现拼接错误的情况。
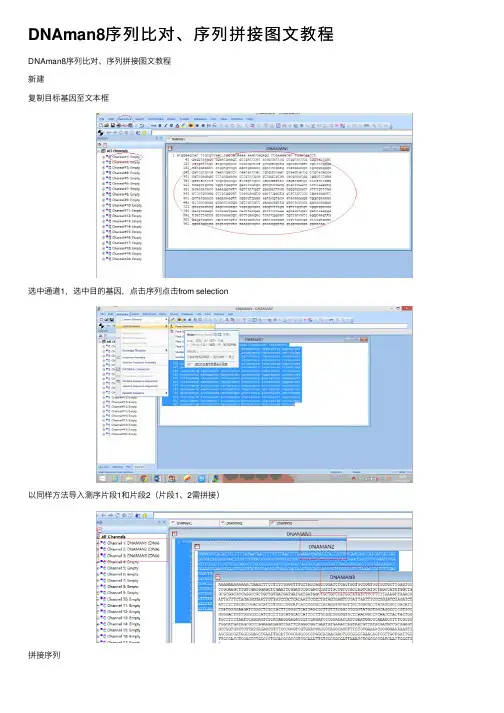
磊 DHAStar步骤2:N ewEdi tSeq GeneQuest MapDr awMegAli gnFrimerSelect ProteanCtrl+OCtrl+N (Jni: ii ledCout 1 €Save Sas..,Au. t o S 注H E .- …S ave W i nLdow..亠 * Ctrl+SExp or t Revear 1LFir oj ec t T .,Fr i rtt S T*r i it t...E t_qp.亠 * C^rl+F R ◎亡on .七 D o cnmerLt s ►C±rT+WE H i t en L □ L Imp or t M &ir ge.. Cl aww步骤3: File Edit Sequsric^ Contig Projset Search Htlu Can't find thp vector rst A L GE VFCTOKS,VCJ, T - - -「?] X |nDiEl^直找范围⑴:|」新建文件夬 □ 3-11己)Tt ■仙址d T«xt?il«E 口新連文件吏 文件名(B ):左件类型⑴ ACTORS VCT 用CTORS.VCrl 帮助曲Seqlan1 电 Elit S 已guM 亡电 CoMig Project Nd Search Keljt 点击打jrCflnilirt^NP SpitSet Ends D iiwsO步骤5:Set EridsnTom EndsDLinits Vector VectorJTrim Ends進彈.点中广 Quality [50~障改却3*End直击选择Scan Sdectrons匚Untitled□E®I KJ Un ass eab led Sequences n x500修Set Vector ^1 | Set Vector5" End "込 Ihdxpi :祝!L*g1Verto-r 3E ATI : ending Opc ■込!Oldkf.^>eridhTi<g ItqutnECJ : 0nit LimitsVtCMEVecwrJ:m ErdsDAssembfleTria Eb~idj : pending Vs ctr&r 3txn pending C 的昨3 E <aji :Dpt-imize Orde r: pendirtgStgy 电巧UH ;*:・ File Scan AllNameLengthOptions :口JnasseRbledSequences_n XAE»imb|a| jAdd SequenicesD |S et &血口| | Trim 日1血口| Options □Tri m End J : pEndin^Set Ve ctor T | Set Vector▼IAO7 CB100401?Oe 7031.,霸挟轰到的文件点中后,施拉到此界面即可步骤7 Seqlait F02_CB10033S040_0267... D04 CB100329D40 0266.步骤8Ediiti < Frfljset Met S«urch HtlpKcp nrt □回冈 IJl»UhSpetifi^d Senfth 11 HTOP.IMG FO 2_ra 100329040_0 26? t pgea-2-1 - sp 6 - ah 1 (1> 900 > in Contig 1:j*- rrERiw& DQ C CB LGO 3^9D4O 2Q 1. tT 2g. ai 1( x>900 ] in conug :— .*psed Tlbe ChB : 5 Untit led严J窗贞壬4Tfi=打看c a «d_的料C^nffc?W$NP $CSedan:Optiini se Orde r ■ pendlirb9 3 « quenr E JF ; 1FileLimitsVectorVector捜索结果回区文件的 编辑(I 〕查看(V)坝藏如 TMC出址⑴J 搀索结果搜隶D04 CB100329040 026S. pgem-2-1. t?2€. abl F02_CE10032g04Q 0267, pzern-Z-1 sp&. abl” F!rm11P KH iArt Net Searcli Help点击拼搖B叵 1区Assemble/ Aid Sequences □Set &>df □Trim End 呂口1TrimVector Y E :xriip^Ti^ding C壮s :Opt-ihhi se Orde r : p ejn>di.ng号口昭m 匚*苗;2FileLin il:sSet VectorSet VectorVectorVector帮助CHj文f点新運00要搜索的文件或文件夹名为W:D04,CB10032W40_0266. pgem-2-l_ tTZg. seq R搜素文件和文件夹 File EditAO 7 CB10040170e 7031_.Option 生 口|Z'DOO\7也Positions 12・042kb40I,, ・・TtiarulateConsenJusCTCCGGC CGCCATTGGCGGC C GC :GGGAATTC(J ATTCCGCCTGTTCGTCGGC命D04_CB100329040_0266.pgeu-2-1 ■ t7Zg . &B1 (L>300)J 回区I|M Strategy of Unlocated Contigs UNG FO 2=CEIOO 323040^0Z67.pgea-2-i.sp6. aJbi (1>9DO )m contig 1; |UNG DO 42C E1OO 3 Z^O-MJ^Q 266. p gen-2 -1. t?2g a Hi 1 (1>9 0 0) in Contig :_•T~iTTiiP 门店「I 2 %匚mNCoverage厂 Conflicts Contig 1D04_CB 100329040_026&.pgeM-2-l. t72g.abl(l>900)A07_CB100401706_7031.pgeii-2^1.WlFC1004017002m abl (l>900) F02 CB100329040 0267.pgen-2^1.sp 6.abl i1>900)步骤io : unlocated Conti CTSoJ ContJE I1□ © xn Ml临丁 HCGC CGCI :辽站UGG€ I :丸GGCAATTCGATTtXGCtTgTTCGTl : GGC弓 SeqKan File Edit Sequence Conli i 4 Praiject 1 fiet Search. KelpSunni ary SUtisticE Rep QF tTrio R 电FQT tG-eneita c CodkEdit Sflltcttd Cod*WTERIIG F P SEQ(l>20) i lapsed Tiiae 0^0:0 g«| |»Uhfpttilitd SeanhAfse-nihlin";WlFHTOffWSc^FBic-erit agoAssembling# Ajseniihliiifi 中不奋迭澤设査 MatehSce■■盯■hid T 區. 巨I Assemblri Enply lapijrt … Inply Irin B»pprt. •Up a ⑷►Cre BL I Q Higw Gr^upCtrl+:QrdaF CQstigsY HC ^Q F Latal^g... Cpnlnni^ant StQE... Repala 13 vt Stqs... ConserKus L.aiinQ S ViewingDuabEnd Specifier Canflict/SNP Split Editing & Cola GM 四 Seivftii kernelMmum Sequence Length AdvancedMaximumi 氏dded Gaps per kb irn CcnligMawmum Added Gaps per kb n Sequence[70~Regisler Shift Dilfeienee丁L 胡 group CamtdefBd1=- Gap Penatii low -Gap Length Penaky 设亨氓毕后卓击|o.?oHelp步骤11: 按照步骤6将PCR 特异性引物序列放进文件中,与测序结果和目的序列等进行比对。
分子定点拼接
分子定点拼接是一种通过PCR等方法向目的DNA片段中引入所需变化的分子生物学技术。
这种方法可以用于改造基因,改变功能序列的编码特性。
定点拼接技术广泛应用于基因调控因子、DNA和蛋白互作、蛋白结构和功能、酶学活性位点的确定等领域。
在定点拼接过程中,首先需要设计引物,使目标DNA分子的5'端与3'端具有同源互补序列。
然后,通过PCR反应将两个DNA片段拼接在一起。
在退火延伸过程中,DNA聚合酶会沿着暂时桥接上的长DNA单链补全碱基,从而形成完整的拼接后DNA 双链。
一旦一个拼接好的DNA分子形成后,只有拼接DNA末端引物能够继续进行PCR反应,而单独扩增出的DNA小片段会作为引物扩增出拼接DNA。
这样,两段DNA序列就会拼接到一起并扩增。
分子定点拼接的应用之一是在载体中插入DNA片段。
具体方法为,首先使用合适的正反向引物对DNA插入片段进行PCR 克隆,得到最终的PCR产物,产物两端的序列与载体重叠。
然后将载体和片段混合,通过变性退火使插入片段与载体杂交并在Phusion DNA聚合酶的驱动下延伸。
几轮PCR循环后,得到的产物是带有两个缺口的融合质粒(每条链一个)。
总的来说,分子定点拼接是一种有效的DNA拼接方法,它
不需要限制酶位点即可连接DNA分子,且准确率高,应用广泛。