喹诺酮类抗菌药分类、构效关系.
- 格式:doc
- 大小:378.00 KB
- 文档页数:3

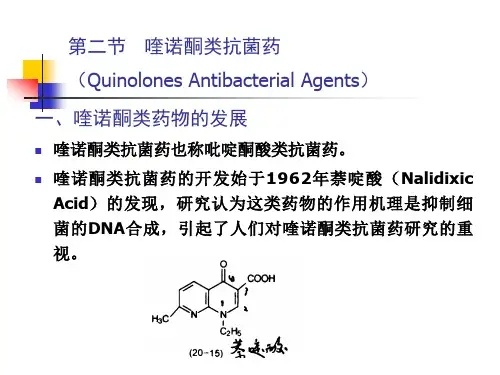
人工合成抗菌药一.喹诺酮类(quinolones)是含有4-喹诺酮母核的抗菌药,此类药物发展迅速,新喹诺酮类是6位引入F,7位引入哌嗪基或其它基团的衍生物,称为氟喹诺酮类,具有抗菌作用强、抗菌谱广、口服吸收好、组织分布广、毒性低等优点,因而成为广泛应用的药物。
氟喹诺酮类药物共性1).抗菌广,抗菌作用强,对G-杆菌包括绿脓杆菌、G+菌金葡菌及产酶金葡菌均有强大作用,某些药对结核杆菌、支原体、衣原体及厌氧菌也有效。
2).抑制细菌的DNA螺旋酶,使DNA复制受阻,导致DNA降解及细菌死亡。
由于其独特的抗菌作用机制,故与其它抗菌药间没有交叉耐药性。
耐喹诺酮的细菌染色体突变使DNA螺旋酶改变、膜孔蛋白通道改变或药物泵出机制被激活而耐药。
3).口服吸收好,蛋白结合率低,体内分布广,组织体液内浓度高。
半衰期较长,多经尿排泄。
4). 适用于敏感菌所致的泌尿道、胃肠道、胆道、呼吸道、前列腺炎、淋球菌性尿道炎或宫颈炎、骨、关节、皮肤软组织等感染。
5).不良反应少而轻,常见恶心、呕吐、皮疹、头痛、眩晕、失眠等症状,大多数停药后即可恢复。
2.常用喹诺酮类药物二. 磺胺药是氨苯磺胺的衍生物,氨苯磺胺分子中的磺酰胺基上一个氢原子(R1)被杂环取代可得到一系列抗菌作用和体内过程存在明显差异的磺胺药如磺胺嘧啶、磺胺异唑等。
如将氨苯磺胺分子中的对位氨基上一个氢原子(R2)取代则可得到口服难吸收的用于肠道感染的磺胺药如酚磺噻唑等。
磺胺药分类:1. 用于全身感染的磺胺药根据磺胺药口服吸收的难易程度和应用部位不同可分为三类:短效类(t1/2<10h):磺胺异唑(SIZ)中效类(t1/2在10~24h):磺胺嘧啶(SD)、磺胺甲基异唑(SMZ)长效类(t1/2>24h):磺胺对甲氧嘧啶(SMD)、磺胺邻二甲氧嘧啶(SDM')2. 用于肠道感染的磺胺药磺胺脒(SM)、酚磺胺噻唑(PST)、柳氮磺胺吡啶(SASP)3. 外用磺胺药磺胺醋酰(SA)、磺胺嘧啶银(SD-Ag)、甲磺灭脓(SML)药理作用抗菌谱较广。

喹诺酮类抗菌药的构效关系及不同成酸盐的原因在我们的日常工作中经常会遇到诸如盐酸左氧氟沙星、乳酸左氧氟沙星、甲磺酸左氧氟沙星等喹诺酮类药物的困惑,且在论坛上多次被大家所提及。
本人以微薄之力,短见浅识地向大家介绍为什么会出现如此让人摸不着头脑的问题。
在此首先要万分感谢广州医科大学有机化学教研室陈老师、临药网clinphar老师、丁香网药物化学讨论版天使猪及其他老师的指导,正是在他们的引导下我从自以为通天彻地地撰写了这篇文章到发现重大技术错误到现在才可以有脸出来。
本文涉及有机化学、药物化学等内容,对化学头晕乏力,心悸气促的网友们大可以直接阅读表格后的一段。
首先在了解这个问题前大家应该先了解喹诺酮类抗菌药的构效关系和有机酸的部分理化性质和结构特性。
(一)、喹诺酮类抗菌药的构效关系喹诺酮类抗菌药作用机制主要为双重抑制拓扑异构酶Ⅱ(DNA 旋转酶)和拓扑异构酶Ⅳ,从而影响DNA的正常形态与功能达到抗菌目的。
DNA是以高度螺旋卷紧的形式存在于菌体内,如果不卷紧,则其长度远远超过细胞壁,根本无法容纳在细胞壁中,也无法进行正常的DNA复制、转录、转运与重组。
DNA螺旋酶的作用就是使DNA保持高度卷紧状态。
喹诺酮类药物妨碍此种酶,造成染色体的不可逆损害,而使细菌细胞不再分裂。
拓扑异构酶Ⅳ在细胞壁的分裂中对细菌染色体的分裂起关键的作用。
喹诺酮类药物的毒性包括:1.极易与金属离子形成螯合物,所有喹诺酮类药物的一个共同特性是与金属离子特别是二价金属离子形成螯合物,不仅降低了药物的抗菌活性,同时也使体内的金属离子流失,尤其对妇女,老人和儿童能引起缺铁、缺锌、缺钙等症。
这方面内容可以翻阅《有机化学》路易斯酸碱理论及配合物等相关理论。
2.光毒性;3.药物相互反应(与P450);4. 软骨毒性。
5.有少数药物还有中枢毒性,胃肠道反应和心脏毒性。
这些毒性都与其化学结构有关。
1、A环是抗菌作用的基本结构,变化小;B环可作较大的改变,可以是苯环、吡啶环、嘧啶环等。


1 品种及作用喹诺酮类药物化学结构构效关系:1、喹诺酮母核的3位均有羧酸基,6位引入氟原子可增强抗菌作用并对金葡菌有抗菌活性。
2、7位引进哌嗪环可提高对金葡菌及绿脓杆菌的抗菌作用(如诺氟沙星),哌嗪环被甲基哌嗪环取代(如培氟沙星),则脂溶性增加,肠道吸收增强,细胞的穿透性提高,半衰期延长。
3、在8位引进第二个氟原子,可进一步提高肠道吸收,延长半衰期(如洛美沙星等)。
4、N-1修饰以环丙基团(环丙沙星)或噁嗪基团(氧氟沙星)可扩大抗菌谱,增强对衣原体、支原体及分支杆菌(结核杆菌与麻风杆菌等)的抗菌活性,噁嗪环还可提高水溶性,使药物在体内不被代谢,以原形经尿排泄。
分代及特点:第一代 萘啶酸、 吡哌酸对G -杆菌作用强,仅适用于尿路、肠道感染第二代 诺氟沙星、氧氟沙星、环丙沙星对G -菌作用强,体内较稳定,毒性降低,可用于各系统感染第三代 左氧氟沙星 、依诺沙星、氟罗沙星、洛美沙星、司帕沙星、格帕沙星在二代基础上增加了对G +球菌、衣原体、支原体、军团菌和结核杆菌的作用,安全性高,半衰期长第四代 莫西沙星、加替沙星 、克林沙星在三代基础上增加了对抗G +球菌的活性,增加了对厌氧菌的抗菌活性 抗菌作用:1、喹诺酮类药物对肠杆菌科细菌具有强大的抗菌作用,以环丙沙星为最高,左氧氟沙星和氧氟沙星次之;对不动杆菌和铜绿假单胞菌的抗菌作用较肠杆菌科细菌差;流感嗜血杆菌呈高度敏感,奈瑟氏菌属多呈敏感。
X N O OH O F R7R1R2R5R82、喹诺酮类药物对G+球菌亦具有抗菌作用,但其抗菌作用明显较肠杆菌科细菌低,以左氧氟沙星相对最高,环丙沙星和氧氟沙星略低;仅对金黄色葡萄球菌(除甲氧西林耐药外)具抗菌活性。
3、对衣原体、支原体、军团菌和结核分支杆菌及其他分支杆菌具有一定作用。
药动学特性:1、多数品种口服吸收良好,血药浓度相对较高。
2、半衰期较长,多在3-7h。
3、蛋白结合率低,大多为14%-30%。
4、体内分布广泛,组织和体内浓度常高于或等于血药浓度,可达有效治疗水平。

喹诺酮类抗菌药物• 分类• 作用机制、化学结构和构效关系• 抗菌作用、耐药性• 氟喹诺酮类抗菌药的药理、不良反应、临床应用及注意事项历史近10年来发展十分迅速的抗菌药物,国内普遍应用,有许多优点,也有很多不足。
目前滥用严重,耐药菌株增多,尤其大肠杆菌等。
发展阶段第一阶段:1962年合成萘啶酸,不良反应多,已经淘汰。
第二阶段:1974年合成吡哌酸,对G-杆菌作用强,适用于尿路、肠道感染。
第三阶段:1978年氟喹诺酮类问世。
第四阶段:楚瓦沙星、司帕沙星问世最新分类第一代萘啶酸、吡哌酸对G-杆菌作用强,仅适用于尿路、肠道感染第二代诺氟沙星、氧氟沙星、环丙沙星对G-杆菌作用强,体内较稳定,毒性降低,可用于各系统感染第三代左旋氧氟沙星、依诺沙星、氟罗沙星、洛美沙星、司帕沙星、格帕沙星在第二代基础上增加了对G+球菌、衣原体、支原体、军团菌和结核杆菌的作用,安全性高,半衰期长第四代克林沙星、加替沙星、莫西沙星在第三代基础上增加了对抗G+球菌的活性,增加了对厌氧菌的抗菌活性共同特点:抗菌谱广、抗菌力强、组织浓度高、口服吸收好、无交叉耐药、不良反应少喹诺酮类药物的作用机制• 拮抗细菌DNA旋转酶,干扰细菌细胞的DNA复制而呈现杀菌作用• 主要作用的靶酶:拓扑异构酶II及IV–传统的喹诺酮作用于拓扑异构酶II(DNA旋转酶)–新喹诺酮既作用于拓扑异构酶II ,也作用于拓扑异构酶IV抗菌作用1、氟喹诺酮类药物对肠杆菌科细菌具有强大抗菌作用,以环丙沙星为最高,左氧氟沙星和氧氟沙星次之;对不动杆菌和绿脓杆菌的抗菌作用较肠杆菌科细菌差;流感嗜血杆菌呈高度敏感,奈瑟氏菌属多呈敏感2、氟喹诺酮类药物对G+球菌亦具有抗菌作用,但其抗菌作用明显较对肠杆菌科细菌低,以左氧氟沙星相对最高,环丙沙星和氧氟沙星略低;仅对金葡萄球菌(除甲氧西林耐药外)具抗菌活性3、对衣原体、支原体、军团菌和结核分支杆菌及其他分支杆菌具有一定作用耐药性近几年来,伴随着氟喹诺酮类药物在国内的广泛应用,细菌对该类药物的耐药性呈迅速增长趋势,并且各品种间呈交叉耐药。


一、喹诺酮类构效关系:1、A环是必须的药效团,3羧和4酮为抗菌活性不可少的部分;2、B环可以是苯、吡啶、嘧啶;3、1位乙基及环丙基活性强,环丙基最佳(环丙沙星);4、2位取代活性低;5、5位氨基可增强活性.(司帕沙星)6、6位F改善细胞的通透性;7、7位引入杂环,增强抗菌活性,哌嗪最好;8、8位F、甲氧基或与1位成环,增强活性(左氧氟沙星),甲基、甲氧基光毒性减少二、苯二氮卓构效关系要点.1、3位引入羟基(奥沙西泮)降低毒性,并产生手性碳,右旋体作用强。
2、7位有吸电子基可增加活性,吸电子越强,作用越强,其次序为NO 2>Br>CF3>Cl3、5位苯是产生药效的重要基团,5位苯环的2’位引入体积小的吸电子基团.(如F、Cl )可使活性增强。
4、1,2位拼入三氮唑可提高稳定性,并提高与受体的亲和力,活性显著增加。
5、苯环用生物电子等排体噻吩杂环置换,保留活性。
6、1位取代基在体内代谢去烃基,仍有活性。
三、吩噻嗪类药物的构效关系:以氯丙嗪为先导化合物,对吩噻嗪类进行结构改造。
三方面:1、吩噻嗪环上的取代基:吩噻嗪环只有2位引入吸电子基团时可增强活性。
作用强度与吸电子性能成正比,CF3>Cl>COCH3>H>OH。
2位乙酰基可降低药物的毒性和副作用。
2、10位N上的取代基:母核上的10位N原子与侧链碱性氨基之间相隔3个直链碳原子时作用最强,是吩噻嗪类抗精神病药的基本结构。
侧链末端的碱性基团常为叔胺,也可为氮杂环,以哌嗪取代作用最强。
3、三环的生物电子等排体。
四、μ受体选择性激动剂构效关系1、芳环和碱性叔胺氮原子是μ受体激动剂的必要结构部分,二者通过2个或3个碳原子的碳链相连接。
2、芳环3位酚羟基的存在使活性显著增强。
氮原子上以甲基取代活性好,当N-取代基增大到3~5个碳原子时,如烯丙基(纳洛酮)、环丁基甲基时,由激动剂转变为拮抗剂。
3、μ受体选择性激动剂的药效构象相同,其芳环以直立键与哌啶环相连。


收稿日期:2001-02-15 3通讯联系人 Tel :(0371)7763952 E 2mail :Qulingbo @基金项目:河南省自然科学基金资助项目(9913)文章编号:1005-0108(2001)04-0241-04氟喹诺酮类药物构效关系的研究进展屈凌波13,刘艳1,郭宗儒2(11郑州大学化学系,郑州450052;21中国医学科学院中国协和医科大学药物研究所,北京100050)摘 要:综述了国内外有关氟喹诺酮类药物的构效关系研究,对第四代氟喹诺酮类药物的研究、开发具有一定的参考价值。
关键词:氟喹诺酮;构效关系;综述中图分类号:R914 文献标识码:A 氟喹诺酮类药物是20世纪70年代后期发展起来的第三代喹诺酮类抗菌药,它们的问世,开创了全合成抗感染药物的新篇章。
该类药物因其抗菌谱广、抗菌活性强、使用方便、疗效确切的优势,无论是在研究开发还是临床应用方面均备受重视。
目前已有60多个品种经过临床验证或正在临床试用中,其中诺氟沙星(氟派酸)、依诺沙星(氟啶酸)、氧氟沙星(氟嗪酸)、培氟沙型(甲氟派酸)和环丙沙星(环丙氟派酸)已广泛应用于临床,另外还有洛美沙星、托氟沙星(tosufloxacin )、氟罗沙星、芦氟沙星(rufloxacin )、那氟沙星(nadi 2floxacin )、斯帕沙星(sparfloxacin )和左氟沙星(levofloxacin ),也已批准上市。
但是,目前这些临床药物尚有不足之处,例如对G +菌和厌氧菌作用较弱,作用时间不够持久(目前市售喹诺酮类药物中芦氟沙星(rufloxacin )具有同类药物中最长的半衰期36h [1]),具有中枢视神经系统及光敏等不良作用,与某些药物(如茶碱类及非类固醇类药物)有相互作用。
因而继续开发新的喹诺酮类药物,通过结构改造改善以上局限性成为这类药物研究的重点。
目前,氟喹诺酮类化合物的定性和定量构效关系的研究已有很大的进展,研究结果必将对指导这类药物定向合成,加快新药开发进程起到重要意义。
喹诺酮类抗菌药分类、构效关系
喹诺酮类抗菌药的基本结构为吡酮羧酸类衍生物,综合临床使用的喹诺酮类抗菌药的结构,归纳其基本结构通式如下:
12345678Y
X N 1
COOH
R 2
R 3R 4
5
O A B
该类药物的结构特点是在其基本母核结构上一般1位为取代的氮原子,3位为羧基,4位为酮羰基,5、7、8位可有不同的取代基,第三代、四代喹诺酮类抗菌药6位为氟原子。
喹诺酮类药物按其母核的结构特征可以分为以下四类: (1)萘啶羧酸类(naphthyridinic acids )
N N
2CH 3
H 3C
COOH
O
N N CH 2CH 3
N
COOH
O
F
HN
萘啶酸 依诺沙星
nalidixic acid enoxacin
(2)吡啶并嘧啶羧酸类(pyridopyrimidinic acids )
N N
N 2CH 3
COOH
O
N
N N
N 2CH 3
COOH
O
N
HN
吡咯酸 吡哌酸 piromidic acid pipemidic acid
(3)噌啉羧酸类(cinnolinic acids )
N
N O O
CH 2CH 3COOH
O
西诺沙星 cinoxacin
(4)喹啉羧酸类(quinolinic acids )
N CH 2CH 3
COOH
O
N
HN
F
N
O
F
N
HN
COOH
诺氟沙星 环丙沙星
norfloxacin ciprofloxacin
N
O
CH 3
COOH
F
N
N O
H 3C
N
O
F COOH
OCH 3
H N
N
氧氟沙星 莫西沙星
ofloxacin moxifloxacin
N
O
F
N
HN
COOH
F
3
NH 2
H 3C
N
O
F
N
HN
COOH
OCH 3
3
司帕沙星 加替沙星 sparfloxacin gatifloxacin
在这四类结构中,喹啉羧酸类药物最多,发展最快。
根据喹诺酮类抗菌药的化学结构和抗菌作用的关系,将该类药物的构效关系总结如下: 1.吡啶酮酸的A 环是抗菌作用的基本药效基团,变化较小,其中3位-COOH 和4位C=O 是抗菌活性必需基团,若被其他取代基取代则活性消失。
2.B 环可作较大改变,可以是苯环(X=CH ,Y=CH )、吡啶环(X=N ,Y=CH )、嘧啶环(X=N ,Y=N )等。
3.1位取代基为烃基或环烃基活性较佳,其中以乙基或与乙基体积相近的氟乙基或环丙基的取代活性较好。
4.5位可引入氨基,提高吸收能力或组织分布选择性。
5.6位引入氟原子可使抗菌活性增大。
引入其他不同取代基对抗菌活性贡献的大小顺序为:-F >-Cl >-CN≥-NH 2≥-H 。
6.7位引入五元或六元杂环,抗菌活性均增加,以哌嗪基最好。
7.8位以氟、甲氧基取代或与1位成环,可使活性增加。