制剂车间生产过程风险评估报告
- 格式:docx
- 大小:85.19 KB
- 文档页数:7
制剂车间生产过程风险评估报告
GMP文件
题 目 制剂车间生产过程质量风险评估
编 号 第 1 页 共 页
草拟/ 订正 日 期 拟订部门 制剂车间
审 核 日 期 颁发部门 质量部
批 准 日 期 奏效日期
散发部门 质量部、制剂一车间、制剂二车间、生产部
目的:
对生产的全过程中全部可能出现的风险进行评估,确立各剂型要点控制的目标,核实和拟订纠正和预防举措,关于高风险和中等风险的一定确立降低风险的举措,低风险增强生产
过程控制,保证产质量量,降低风险发生的可能性,提升可检测性(可检测性) ,将风险控制在可接受水平。
将风险评估的结果应用于指导工艺规程、车间管理与操作规程的订正与工艺考证。
范围: 制剂车间生产全过程。
责任: 制剂车间工艺员负责风险信息采集 ,质量部负责审查、同意。
内容:
1 概括
我企业生产销售同意的中西药产品 62 个,常年生产品种 21 个,常年生产的品种主要有:
*** 片等。企业对制剂一车间、制剂二车间、提取车间的厂房、生产设备和设备多产品共用可行性进行了风险评估并采纳了举措,已将交错污染的风险降低为可接受水平。
针对企业实质的生产状况,对制剂车间生产的全过程进行了剖析,对每一工艺过程中可能会发生的影响产质量量的步骤进行风险剖析、评论微风险控制,确立优先控制的目标和推行的举措,降低风险发生的可能性,提升可检测性,将风险控制在可接受水平。
2 风险管理剖析方法
失败模式成效剖析法( FMEA)
3 职责 风险评估小组: 组长:负责风险管理的协调与风险评估文件的撰写。
组员:负责采集和组织风险信息, 提出风险项目, 剖析、评估风险项目, 并提出降低风险项目的举措。
名称 人员
组长 *****
组员 ****** 制剂车间生产过程风险评估报告
4 依照
《药品生产质量管理规范》 ( 2010 年订正)
《药品 GMP指南》
5 风险评估
FMEA摆列标准和失败得分以下:
风险的严重程度
结果 结果的严重性 评分
严重危害 会致使整批产品报废或出现法例风险 5
高 会出现严重误差或致使产质量量出现异样,造成部分报废或致使 4
用户投诉
中等 会出现重要误差或可能会致使产品返工,对产质量量有必定影响 3
低 会出现细小误差,对产质量量影响较小 2
细小 对产质量量无影响 1
风险的发生几率
失败发生的可能性 举例 评分
特别高:几乎不行防止失败 极屡次的发生 5
高:频频发生的失败 每天发生 4
中等:有时发生的失败 每个月发生 3
低:相对特别少发生的失败 每几个月发生一次 2
细小:几乎不行能发生的失败 仅发生过一次 1
风险被检测或发现的可能性
发现的可能性 在发生以前经过过程控制能够检测出缺点的可能性大 评分 小
绝度不行能或极小 完好没有有效的方法或当前的方法几乎不行能检测出 5 失败模式
可能性较低 当前的方法只有较低的可能性能够检测出失败模式 4
中等可能性 当前的方法有中等的可能性能够检测出失败模式 3
可能性较大 当前的方法有较大的可能性能够检测出失败模式 2
可能性特别大或几乎肯 当前的方法能够检测出失败模式的可能性特别大或几 1 定能 乎能够必定,有靠谱的检测方法。
风险指数确实定:
对风险出现的可能性、严重性和可检测性依据上表分别进行打分 , 确立风险指数
RPN=出现的可能性×严重性×可检测性
风险级别:
得分 1-25 分为低风险, 26-59 分为中等风险, 60-125 分为高风险。 RPN为 20 分以下为可接受风险。
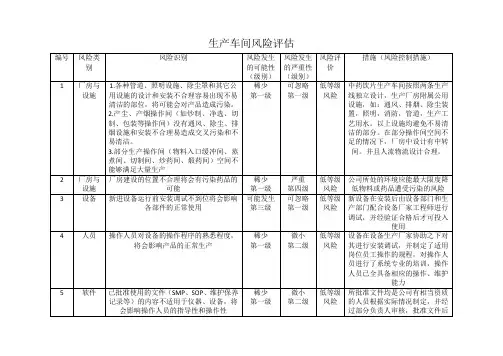
制剂车间生产过程风险剖析与控制 制剂车间生产过程风险评估报告
风险评估与控制及工艺考证项目确认
风险剖析
风险项目 风险描绘 已有举措 RPN 风险级别 风险接受 能否需进行工艺考证 / 备注 严重性 可能性 可检测性
1. 操作前检查房间压差表、温湿度,压差切合要求后再开始生产,并实时将记
录填写在批记录上及压差记录表上。
房间压差、温湿度不切合规定 3 2 2 12 低 可接受 生产时监控
2. 空气干净级别同样的压差应大于 5Pa 以上,一般区与干净区之间差压应大于
10Pa 以上。
1. 对 D级干净区起码每三个月进行一次动向监测(浮游菌、沉降菌、表面微生
物、尘埃粒子)
2. 改换高效过滤器后应静态监测尘埃粒子、浮游菌、沉降菌、表面微生物,并
微生物、尘埃粒子不切合规定 应依据实质需要进行监测风量风速、换气次数、过滤器压差等项目。 5 2 2 20 低 可接受 生产时监控
环境 3. 系统停止运转必定周期(一般指 3 周)需从头启动后的静态监测项目包含尘
埃粒子、浮游菌、沉降菌、表面微生物。
4. 每年起码 1 次消毒,停产 10 天以上者,动工前需消毒
1. 生产后 QA负责清场管理,保证设备依照预约的洁净规程进行洁净,检查合
格后方可发放“清场合格证” 。
生产完成后洁净不完全 4 1 2 8 低 可接受 生产时监控
2. 每年 9-11 月进行车间操作、卫生、工艺等脱产培训。
3. 各工序均有明确的洁净规程,并有明确的QA检查标准。
产尘工序尘埃污染干净走廊,导1. 拟订 “ 防备尘埃产生和扩散的管理程序 ”规定各工序操作时减少尘埃产生的
4 2 1 8 低 可接受 生产时监控
致交错污染 方法以及产尘工序清场要求并规定操作间内需保持相对负压。
1. 职工的 SOP培训,保证按规定进行操作。 3 2 2 12 低 可接受
未按要求进行洗手、换衣,裸手 平时检查,随时纠正。
生产时监控
人员 直接接触药物 3. 拟订了“人与物料、中间品、半成品接触的标准操作程序” ,规定了不得裸手
接触药物。
培训不到位,不可以有效履行文件 1. 获得上岗证前不得独立操作 4 2 3 24 低 可接受 生产时监控 / 考证时依据方 制剂车间生产过程风险评估报告
规定 2. 班长、 QA平时检查,随时纠正。 案要求进行适合培训,操
3. 每年 9-11 月进行车间操作、卫生、工艺等脱产培训。 作应切合考证方案要求
1. “干净区领料标准操作程序”和工艺规程中均规定了所领物料一定检查能否 5 1 2 10 低 可接受
有合格的查验报告单
2. “原、辅料发放标准操作程序”规定发放的物料一定有合格的报告单、放行
采纳过期、超出复检期的物料 单 生产时监控
,且发料前要查对物料能否在复验期内。
3. 放行单规定有有效期,防止库房人员误将过期 / 过复验期的放行单当成合格的物料发出。
物料出入 D级干净区无人管理, 1. 拟订了《物料出入 D级干净区的标准操作程序》规定了物料进入缓冲间一定 4 2 1 8 低 可接受
致使 D 级干净区环境污染,物料 先用吸尘器吸净并脱去外包,做好物料标示,其实不得同时开启缓冲间的两门 生产时监控
物料 混入异物。
1. 拟订了“周转容器标准管理程序”规定周转桶推行专人专职管理 4 1 3 12 低 可接受
周转桶管理杂乱,致使交错污染 2. 拟订了“容器内、外标记操作规范与操作程序”规定周转桶领用前一定查对 生产时监控
洁净状况及有效期,规定标示不得直接接触物料且桶内外各放一张表记
不行利用物料管理杂乱,致使不 1. 拟订了 “不行利用物料办理标准管理程序 ” 规定了不行利用物料的根源且不 5 1 4 20 低 可接受
生产时监控
可利用物料混入 可再利用,一致交于中间库管理员管理,并一致报废办理。
1. 拟订了 “物料均衡标准管理程序 ”规定了物料均衡计算的公式及高出范围的 5 2 1 10 低 可接受
物料投入与产出不均衡,出现混
办理方式 生产时监控 / 考证时确认
料的状况
2. 各工艺规程规定了各工序物料均衡范围
压缩空干净度不切合要求 每三个月对压缩空气的干净度进行平时检查。 4 2 2 16 低 可接受 生产时监控
设备、设备及厂房超出确认有效 每年拟订考证总计划,确认全部仪器设备、设备在考证有效期内 3 3 1 9 低 可接受
考证时确认
期
设备、设
设备破坏,带病生产致使产品不 成立了《设备保护、养护得管理程序》及《预防性保护计划标准管理程序》 ,以
施及厂房 生产时监控
合格率上涨 保证设备在破坏前获得有效保护,保证不带病生产
计量用具未经校验或超出校验有 1. “度、量、衡用具标准管理程序 ”规定了用具计量管理制度,计量用具一定 5 3 1 15 低 可接受 生产时监控 制剂车间生产过程风险评估报告
效期或称量范围不合用,称量不 经校准后贴上检定合格证后方可交予车间使用,每年对车间内使用的胸怀衡器
正确致使投料错误 拥有专业人员检定一次。
2. “计量、仪器管理的标准程序 ”规定了生计科负责一致管理企业计量用具,
各部门建立计量员
3. “计量用具周期检定标准管理程序 ”对个用具进行分类管理并规定各种用具
检定周期
4. “车间生产过程复核标准管理程序 ”对投料、计算、称量、中间站出入等内
容都规定要按文件要求进行复核,保证操作正确。
1. 工艺规程中规定操作前需检查确认洁净状况,操作后需按要求达成洁净 5 1 3 15 低 可接受
设备设备等洁净不完全污染药品 2. QA及工艺员按要求进行核查,详见“现场 QA岗位职责”,“工艺员岗位职
责”
细度达不到要求,保证不了药物 1. 工艺规程中拟订规定目数要求 3 3 2 18 低 可接受
预办理 和辅料的混淆平均性以及适合的 2. QA及工艺员在生产前、后对筛网的完好性进行核查,详见“现场 QA岗位 生产时监控 / 考证时确认
溶出速度。 职责”,“工艺员岗位职责”
制粒机超出其最大的承载量,导 1. 工艺规程中规定详细的每锅投料量、制粒及整粒筛网目数、出料颗粒水分 5 2 2 20 低 可接受
致颗粒含量不平均 总混时间
混淆搅拌时间及制粒搅拌时间不 2. QA及工艺员按要求进行核查,详见“现场 QA岗位职责”,“工艺员岗位职
够,致使颗粒含量不平均 责”