转录组Denovo手册(无答案)
- 格式:pdf
- 大小:2.01 MB
- 文档页数:23

图1 10X Genomic linked-reads辅助基因组组装流程图表1 不同组装策略组装人的基因组大小和ScaffoldN50长度[1]随着技术的发展,越来越多的物种完成了基因组的测序工作。
但基于二代测序短读长的限制,制约了参考基因组的组装质量,从而影响了后续研究工作的开展。
如今,我们可以利用更多的新技术,如10X Genomics,BioNano,ChiCago等,将基因组组装结果进行完善,进一步构建出高质量的参考基因组。
10X Genomics linked-reads10X Genomics公司通过在序列中引入barcode序列,能够得到跨度在50-100Kb的linked reads信息,与二代测序数据相结合,在Scaffold 的组装上能够得到媲美三代测序的组装结果(表1)。
展开阅读10X Genomic linked-reads辅助基因组组装流程如下图所示:图2 光学图谱工作流程图表3 利用Chicago技术提升相应的指标图3 Chicago文库构建流程图[6]Chicago文库构建流程如下:基因组 de novo 组装新技术助力文章冲刺新高度[1] Mostovoy Y, Levy-Sakin M, Lam J, et al. A hybrid approach for de novo human genome sequence assembly and phasing[J]. Nature methods, 2016. 阅读原文>>/nmeth/journal/v13/n7/abs/nmeth.3865.html[2] Pendleton M, Sebra R, Pang A W C, et al. Assembly and diploid architecture of an individual human genome via single-molecule tech-nologies[J]. Nature methods, 2015. 阅读原文>>/s?wd=paperuri:(ac8d0768*******de9b67e959e5d924b)&filter=sc_long_sign&sc_ks_para=q%3DAssembly+and+diploid+architecture+of+an+individual +human+genome+via+single-molecule+technologies.&tn=SE_baiduxueshu_c1gjeupa&ie=utf-8&sc_us=14004045691020250024[3] VanBuren R, Bryant D, Edger P P , et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum[J]. Nature, 2015. 阅读原文>>/s?wd=paperuri:(4f4baa5f458c3598ebfa32b1017a4569)&filter=sc_long_sign&sc_ks_para=q%3DSingle-molecule+sequencing+of+the+desiccation-tolera nt+grass+Oropetium+thomaeum.&tn=SE_baiduxueshu_c1gjeupa&ie=utf-8&sc_us=3671601047694710580[4] Dong Y, Xie M, Jiang Y, et al.Sequencing and automated whole-genome optical mapping of the genome of adomestic goat (Capra hircus). Nature biotechnology, 2013, 31(2): 135-141. 阅读原文>>/nbt/journal/v31/n2/full/nbt.2478.html [5] Zhang Q, Chen W, Sun L, et al. The genome of Prunus mume. Nature communications, 2012, 3: 1318. 阅读原文>>http://pubmedcentralcanada.ca/pmcc/articles/PMC3535359/[6] Bredeson J V, Lyons J B, Prochnik S E, et al. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity[J]. Nature biotechnology, 2016, 34(5): 562-570. 阅读原文>>/s?wd=paperuri:(030555bb483ea9f72bf308bf22787f02)&filter=sc_long_sign&sc_ks_para=q%3DSequencing+wild+and+cultivated+cassava+and+related +species+reveals+extensive+interspecific+hybridization+and+genetic+diversity.&tn=SE_baiduxueshu_c1gjeupa&ie=utf-8&sc_us=13838504648880517513[7] Putnam N H, O'Connell B L, Stites J C,et al. Chromosome-scale shotgun assembly using an in vitro method forlong-range linkage[J]. Genome research, 2016, 26(3): 342-350. 阅读原文>>/s?wd=paperuri:(4c8ec46542c7e21bfa15ae10f7a9f8bf)&filter=sc_long_sign&sc_ks_para=q%3DChromosome-scale+shotgun+assembly+using+an+in+vit ro+method+for+long-range+linkage.&tn=SE_baiduxueshu_c1gjeupa&ie=utf-8&sc_us=36575566455777547参考文献Chicago技术(体外Hi-C 技术)作为提供长距离连接数据的组装提升方法,Chicago技术不仅能够获得长序列连接信息,还能帮助组装提升到染色体水平,该技术使用效率高、操作简便、经济性强,并且产生的高质量文库能够更好地应用于后期组装或研究。

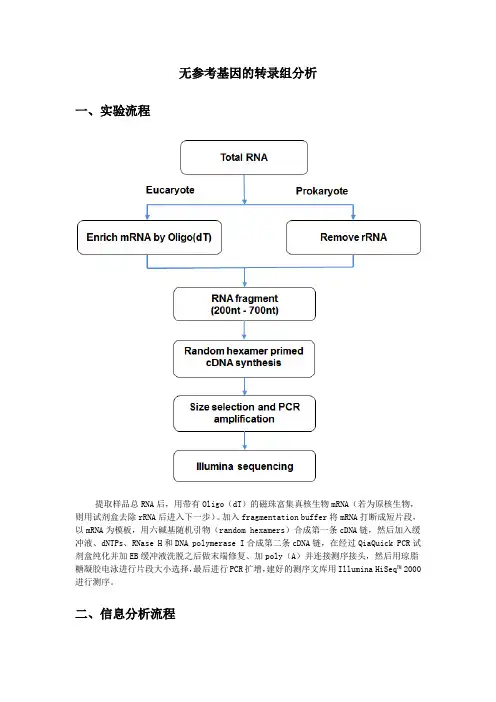

动植物基因组de novo常见问题基础知识1、什么是基因组de novo测序答:对某一物种进行高通量测序,利用高性能计算平台和生物信息学方法,在不依赖于参考基因组的情况下进行组装,从而绘制该物种的全基因组序列图谱。
2、普通基因组的定义答:单倍体,纯合二倍体或者杂合度<%,且重复序列含量<50%,GC 含量为35%到65%之间的二倍体。
3、复杂基因组的定义答:杂合率>%,重复序列含量>50%,GC含量处于异常的范围(GC 含量<35%或者GC含量>65%=的二倍体,多倍体。
诺禾致源对二倍体复杂基因组进一步细分为微杂合基因组(%<杂合率<%=、高杂合基因组(杂合率>%)以及高重复基因组(重复序列比例>50%)。
4、怎么查询基因组的大小答:查询植物基因组大小的网站:;查询动物基因组大小的网站:。
、5、基因组的项目周期6、基因组承诺的组装指标答:简单基因组:contig N50>20K,scaffold N50>500K;复杂基因组:contig N50>20K,scaffold N50>300K。
样品要求1、动植物基因组测序对取样有什么要求答:植物:需要黑暗无菌条件下培养的黄化苗、组培苗,基因组样本量500μg~1mg,越多越好。
选择纯合或杂合度尽可能小的样品(杂合度<%)。
动物:应选取肌肉、血液等含脂肪较少的部位取样,尽量选择同一个体取样,以减少个体差异性对后续拼接的影响。
基因组样本量500μg~1mg,越多越好。
样本的性别决定模式是XY型,则尽量选择雌性个体(XX型),如果是ZW型,则尽量选择雄性个体(ZZ型)。
2、全基因组测序对DNA样本有什么要求答:(1)样品需求量(单次):小片段文库,≥3μg;2Kb~5Kb大片段文库,≥20μg;10Kb~20Kb大片段文库,≥60μg;完成全基因组测序样品DNA量需求约为500μg~1mg;(2)样品浓度:对于小片段文库,≥50ng/μl,对于2Kb~5Kb 大片段文库,≥150ng/μl;对于10Kb~20Kb大片段文库,≥150ng/μl;(3)样品纯度:OD260/280=~;无蛋白质、RNA污染或肉眼可见杂质污染;(4)样品质量:基因组完整。

一、概述二代测序(Next Generation Sequencing, NGS)技术的广泛应用,使得基因组学研究取得了长足的进步。
其中,二代测序denovo流程是利用NGS技术对未知生物样本进行全基因组测序,并在此基础上进行基因组组装和注释的过程。
本文将对二代测序denovo流程进行深入探讨,从数据处理到基因组组装和注释等方面进行详细介绍。
二、数据处理在进行denovo全基因组测序之前,首先需要进行数据处理。
数据处理包括测序数据的质量控制、序列过滤和去除低质量序列等步骤。
在质量控制方面,可以利用软件对测序数据进行质量评估,筛选出高质量的测序数据用于后续分析。
针对测序数据中可能存在的接头序列和低质量碱基,需要进行序列过滤和去除低质量序列的处理,确保后续的组装和注释过程能够得到准确的结果。
三、基因组组装基因组组装是denovo流程中的关键步骤,主要是将测序得到的短序列reads进行拼接,重建成完整的基因组序列。
目前,常用的基因组组装算法包括SOAPdenovo、Velvet、ABySS等。
这些算法能够根据reads之间的重叠信息和kmers的频率进行拼接,得到较为完整的基因组序列。
对于大规模基因组的组装,还可以采用高通量测序技术辅助组装,如mate p本人r测序或二代测序测序辅助第三代测序(Hybrid Assembly)等方法。
四、基因组注释基因组注释是denovo流程中的另一个重要步骤,主要是对组装得到的基因组序列进行基因预测、基因功能注释和通路分析等。
在基因预测方面,可以利用软件对基因组序列进行Open Reading Frame (ORF)预测和基因预测,以确定基因的位置和编码序列。
在基因功能注释方面,可以利用生物信息学数据库和工具对基因进行功能和结构注释,帮助研究人员理解基因的生物学功能和作用。
为了进一步了解基因的生物学功能和相互作用,还可以进行通路分析,探究基因在生物体内的作用机制。
五、应用与发展二代测序denovo流程在生命科学研究中有着广泛的应用与发展前景。


IntroductionSOAPdenovo is a novel short-read assembly method that can build a draft assembly for the human-sized genomes. The program is specially designed to assemble Illumina GA. It creates new opportunities for building reference sequences and carrying out accurate analyses of unexplored genomes in a cost effective way. SOAPdenovo是一种新型的short-read装配方法,可以建立一个de novo组装人l类大小的基因组草案。
这个程序是为装配Illumina测序 short reads特别设计的。
它以一种高效益的方式为建立参考序列和计算出精确的未知基因组创造了新的机会。
System RequirementSOAPdenovo aims for large plant and animal genomes, although it also works well on bacteria and fungi genomes. It runs on 64-bit Linux system with a minimum of 5G physical memory. For big genomes like human, about 150 GB memory would be required.SOAPdenovo虽然也能在细菌和真菌基因组也能很好的运行但它的目标是大的植物和动物的基因组。
它运行在最小内存5G的64位Linux系统上。
像人类的大基因组,大约需要150G内存。

动植物Denovo测序知识⼤讲解⾼通量测序的技术开起我们探索动植物基因组奥秘的步伐,提到动植物基因组测序,这就不得不提⼀个概念——de novo测序。
那么什么是de nove测序呢,它与重测序有什么区别呢?De nove测序中Read、Contig和Scaffold等⼜代表什么呢?De nove测序中为什么要建不同⼤⼩⽚段的梯度⽂库?基因注释⼜是注释哪些内容?各位客官别急,且听⼩编给您细细讲来。
1De novo测序概念De novo是⼀个拉丁⽂,代表从头开始的意思,⽽de nove测序则是指在不需要任何参考序列的情况下对某⼀物种进⾏基因组测序,然后将测得的序列进⾏拼接、组装,从⽽绘制该物种的全基因组序列图谱。
由于⾼通量测序长度的限制,⽬前测序策略是先将基因组打断⼩的⽚段,然后再对测出序列⽚段进⾏拼接,最终得到物种的序列图谱如图1所⽰。
图1 ⾼通量测序模式图2De novo测序与重测序区别重测序概念:重测序是全基因组重新测序的简称,是指是对已知基因组序列的物种进⾏不同个体的基因组测序,并在此基础上对个体或群体进⾏差异性分析。
从概念上来看两者的区别在于de nove测序是对没有参考基因组的物种进⾏测序,⽽重测序是对已有基因组的物种进⾏测序,这只是它们区别很⼩的⼀部分。
从原理上来看de nove测序和重测序最根本的区别在于de nove测序需要对测序得到的Reads进⾏拼接组装,⽽重测序得到的数据则是没有组装的短的Reads序列。
值得注意的是,随着测序成本的降低以及组装算法的改进,de nove测序成本越来越低,⽬前来说de nove测序不只对于没有参考基因组物种进⾏测序,还可以对⼀些特有的亚种、品种以及变种等进⾏测序。
3Reads Conting Scaffold概念Reads:即我们通常说的读长的意思,它是指⾼通量测序平台直接产⽣的DNA序列。
Contig:是指Reads基于Overlap关系,拼接获得的长的序列;Scaffold:是指将获得的Contig根据⼤⽚段⽂库的Pair-end关系,将Contig进⼀步组装成更长的序列;关于三者之间的关系如图2所⽰,注意的是Contig是⽆Gap的连续的DNA序列,⽽Scaffold是存在Gap的DNA序列。

生工从零开始解读转录组测序第二版本书着重介绍了转录组测序的概念、原理及基本方法。
这些年我们已经建立了很多新技术,包括各种抗体技术、限制性内切酶技术等等。
但是,在我们实际应用中发现,上述这些技术都有自身不足之处。
如限制性内切酶在大肠杆菌中只能将单链 DNA 切成双链,而抗体却可以利用一个个的片段特异性地与一个个 DNA 片断结合;限制性内切酶往往难以完全识别一条多链的 DNA,而抗体则具备对大量碱基进行高通量识别的能力……这些不足之处,导致目前限制性内切酶和抗体联用的方案效率低下,没有得到广泛应用。
因此,在细胞内,使用转录组测序,仍然是最好的选择。
转录组学是一门新兴的边缘交叉学科,它集中体现了当今生命科学领域里面最先进的研究手段和思想观点。
它把人类认知水平提升到了一个崭新的层次,为揭示生命活动的奥秘打开了一扇窗户。
同时也带给我们许多启迪:1.在生物医药产业界,转录组学正逐渐取代蛋白质组学,成为主流的研究热点。
2.转录组学作为一项非常精准的检验指标,其意义远超过蛋白质组学。
3.随着转录组学的快速发展,越来越多的疾病被确诊并治愈,这无疑会极大推动整个医疗卫生事业的发展。
4.由于转录组学的出现,让更多的临床试验设计者关注临床试验的伦理问题,促进了临床试验的规范化管理。
5.转录组学还引入了一套新的评价模型——“数据驱动”的评估框架,即以转录组学数据为依托,采用统计学方法,定量描述某一生物学过程或状态的表达情况,再根据预期的目标值,运用统计学软件,进行相应的统计分析,最终获得该生物学过程或状态的表征参数。
6.转录组学为后续的基础研究奠定了坚实的基础,尤其是在癌症早期筛查、药物靶向治疗、药物毒副反应监控、药物安全性评价等方面显示出巨大优势。
7.转录组学的诞生改写了人类对生命的认识历史,掀开了生命科学研究的新篇章!生物信息学是现代分子生物学、系统生物学和计算机科学相结合发展起来的一门综合性边缘学科。
生物信息学是一门研究人类生物学信息(基因、蛋白质)的形式、存储、加工、表达、传递、调控、功能和进化的学科。
来说说一个基因组是怎么来的吧(附:仅自己的看法)1.样品的采集、DNA的提取、上机这一部分就略去吧,体力+操作娴熟活,爆个小料,实验组的少堂兄刚和师姐去大理、丽江、临沧等好几个地方花费10天时间采回了一批样。
据说超累超累得活。
去之前还开玩笑说回来可能不认识他了。
或者极端情况是被野兽带走。
哈哈,可见样品的来之不易了吧?跳过。
此步骤过于血腥暴力。
不再展开:)2.测序的原理我们组采用的是Illumina/Solexa测序,它的基本原理是边合成变测序。
在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。
3.操作流程(注:图片引自Elaine R. Mardis (2008) Next-Generation DNA Sequencing Methods Annu. Rev. Genomics Hum. Genet. 9:387–402)简要的表述一下上图测序的过程:1)测序文库的构建准备基因组DNA---随机变短化为几百碱基或更短的小片段---两头加上特定的接头若为转录组测序RNA片段---反转录---cDNA----片段化----加街头值得注意的是:我们这里片段的大小对于后面的数据分析有影响,可根据需要来选择。
对于基因组测序来说,通常会选择几种不同的片段大小,以便在组装的时候获得更多的信息。
2)锚定桥接带接头的DNA片段---变性---与通道上的引物刑场桥状结构---便于后续扩增3)预扩增添加dNTP 和Taq 酶----固相桥式PCR 扩增---变性---释放出互补的单链--通过--不断循环---获得上百万条成簇分布的双链待测片段4)单碱基延伸测序加入四种荧光标记的dNTP 、DNA 聚合酶以及接头引物进行扩增,在每一个测序簇延伸互补链时,每加入一个被荧光标记的dNTP就能释放出相对应的荧光,测序仪通过捕获荧光信号,并通过计算机软件将光信号转化为测序峰,从而获得待测片段的序列信息。
RNA de novo 应用及分析报告作者:李娜娜部门:技术支持部日期:2014-05-16g y n e r g y G e G e n e r g y G e n e r G e n e r g y G e n e r g y G e n e r g y G e n e r g y G G e n e r g y G e n e r g y G e n e n e r g y G e n e r g y G e n e r g e r g y G e n e r g y G e n e r g y g y G e n e r g y G e n e r g y G e n e r g y G e n e r g y n e r g y G e n e rr g y G e G目录一、测序流程2二、案例介绍三、报告解读g y n e r g y G e G e n e r g y G e n e r G e n e r g y G e n e r g y G e n e r g y G e n e r g y G G e n e r g y G e n e r g y G e n e n e r g y G e n e r g y G e n e r g e r g y G e n e r g y G e n e r g y g y G e n e r g y G e n e r g y G e n e r g y G e n e r g y n e r g y G e n e rr g y G e G3RNA de novo 测序:对不依赖任何参考基因组的物种进行转录组测序。
2×100bp ,20~30M reads/sample 。
一、实验原理RNAseq-C01测序数据质量统计RNAseq-C02RNA-seq de novo 组装与统计RNAseq-C03编码蛋白框(ORF/CDS )预测RNAseq-C04Unigene 注释(GO 、KEGG 、COG 、蛋白结构域等)RNAseq-C05SSR 预测分析RNAseq-C06基因差异表达(两个样本以上)其它g y n e r g y G e G e n e r g y G e n e r G e n e r g y G e n e r g y G e n e r g y G e n e r g y G G e n e r g y G e n e r g y G e n e n e r g y G e n e r g y G e n e r g e r g y G e n e r g y G e n e r g y g y G e n e r g y G e n e r g y G e n e r g y G e n e r g y n e r g y G e n e rr g y G e G均一化cDNA 文库均一化cDNA 文库:通过特定处理降低高丰度的cDNA ,使得表达基因对应的cDNA 拷贝数相等或接近,以在后续的测序分析中得到的转录本更加完整,获得更多的转录本信息。
首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们组装变异检测分析,是指对物种基因组进行组装,用组装后的序列进行变异检测分析。
从而得到更多、更准确的SNP,InDel,SV,CNV,Novel gene等变异信息。
组装变异检测对物种中高复杂的结构变异,具有非常高的灵敏性,能够更充分的挖掘出与农艺性状相关基因的变异,为开发相应的分子标记和功能基因研究奠定基础。
技术路线10X Genomics助力组装变异检测升级更全面的SV检测案例解析Linked Reads可有效提供跨度超过几十Kb甚至更长的信息,通过Barcode Overlap关系能够准确的区分单体型变异中不相邻的区域(图3)。
对人NA12878进行检测,通过Linked Reads发现6号染色体中存在一段长达70Kb的缺失(图4)。
同时,对人NA12878中以往发现的8个SV进行Phasing,发现在8个SV中,5个含有较高的Phasing值,3个具有较低的Phasing值(表2)。
对SV(已检测)进行孟德尔遗传分析,发现在NA12878(Mother)中的5个较高Phasing值的SV有3个遗传到了NA12882(Child),进一步研究发现当NA12882存在Deletion时,即对应相关的单体。
此外研究还通过探针序列验证了Phasing值较高的SV的准确性。
而对于Phasing值较低的3个SV,两个通过探针序列验证是假阳性,一个验证是确实存在SV,之所以分值较低是因为这个Deletion在三个个体中均存在(Father、Mother和Child)。
此结果说明利用Linked Reads检测大片段的SV具有较高的准确性。
图4 通过Linked Reads确定6号染色体存在70Kb的缺失表2 Linked Reads对8个SV进行PhasingChr.Location Phase blockBCsintersectingHap1BCsintersectingHap2P DeletedhaplotypeDeletioninheritedbyNA12882Phasingconsistentwithinheritance 1189704509–18978335950188502070574 4.90 × 10–131No YesYesYesYesYesYesYesYesYesYesYes3162512134–162626335190161712492257 6.10 × 10–1525104432113–10450367327098341819528 6.60 × 10–52678967194–79036419*310764666274577.00 × 10–81No678967194–79036419*31078967164502 6.10 × 10–131No839232074–39387229*39030957010450 5.70 × 10–141839232074–39387229*390393708004601599400881–997150152709834181997870.51FP No N/A1437631609–377712286203378120528Common Common N/A14106932640–10717493164010669287010130.68FP No N/ATwo breakpoints of the deletion are on different phase blocks.Low-scoring SV candidates2.80 × 10–140.11图3 通过Barcode Overlap关系可有效检测NA12878存在的结构变异78765000791650007876500079165000NA12878, Chr. 6: 78967194–79036419Barcode overlap013barcodes78765000791650007876500079165000NA12882, Chr. 6: 78967194–79036419Barcode overlap013阅读原文>>。
转录组测序今天我们来学习一些关于转录组测序的知识,从转录组的一些基本概念开始。
第一章Intron:内含子,间隔存在于真核生物细胞DNA中的序列,转录时存在于前体mRNA中,通过剪接过程被去除,最终不存在成熟的mRNA中。
Exon:外显子,真核生物DNA中的序列,与Intron对应,序列在剪接过程中不被去除,最终存在于成熟的mRNA分子中。
UTR:Untranslated regions,非翻译区,信使RNA分子两端的非编码片段。
5'-UTR从mRNA起点的甲基化鸟嘌呤核苷酸帽直至AUG起始密码子,3'-UTR从编码区末端的终止密码子直至PolyA尾的前端。
CDS:code DNA sequence,基因编码区域,mRNA序列中编码蛋白质的序列,以起始密码子开始以终止密码子结束的片段。
转录本(Transcript):基因通过转录形成的一种或多种可供编码蛋白质的成熟的mRNA。
可变剪切:从同一个mRNA前体出发,通过不同剪接方式、选择不同的剪接位点,产生不同的mRNA剪接异构体的过程,可以产生多个转录本。
融合基因:来自不同基因的外显子组合形成新的mRNA,最终产生与外显子来源基因表达产物不同的蛋白质。
start codon,起始密码子;stop codon,终止密码子转录组(Transcriptome):特定生物体在某种状态下所有基因转录产物的总和。
链特异性文库:鉴定真核生物的反义转录本或原核生物的ncRNA。
合成第二链cDNA时用dUTP代表dTTP,使得第二链cDNA上布满含dUTP的位点,然后用特异性识别尿嘧啶的酶消化第二链,得到只包含第一链cDNA信息的文库。
转录组:转录组被测序的物种已经有一个参考基因组。
在分析数据时,不需要拼接转录本,只需要将转录组测序数据与参考基因组进行比较,就可以确定每个基因的表达水平。
无转录组:转录组被测序的物种没有参考基因组,因此需要拼接转录组数据以获得样品中的转录本信息,然后对这些拼接的转录本进行功能注释,然后将转录组数据与拼接的转录本进行比较并计算其表达水平。
【分享】目前最好最完整的SOAPdenovo使用说明转载于泛基因/article/26这是一份关于基因组组装软件SOAPdenovo的使用说明,内容包括了程序使用、参数的详细说明、参数如何调整、各个主要输出文件的格式说明等。
简介:SOAPdenovo(目前最新版是SOAPdenovo2)是利用一种新的组装短read的方法,它以kerm为节点单位,利用de Bruijn图的方法实现全基因组的组装,和其他短序列组装软件相比,它可以进行大型基因组比如人类基因组的组装,组装结果更加准确可靠,可以通过组装的结果非常准确地鉴别出基因组上的序列结构性变异,为构建全基因组参考序列和以低测序成本对未知基因组实施精确分析创造了可能。
程序的下载及安装:下载地址:安装:(a) 下载SOAPdenovo的压缩包(b) 解压缩(c)将得到可执行文件SOAPdenovo和一个配置文件的模板example.contig1 使用程序及参数:SOAPdenovo可以一步跑完,也可以分成四步单独跑一步跑完的脚本:./ SOAPdenovo all -s lib.cfg -K 29 -D 1 -o ant >>ass.log四步单独跑的脚本:./ SOAPdenovo pregraph -s lib.cfg -d 1 -K 29 -o ant >pregraph.log./ SOAPdenovo contig -g ant -D 1 -M 3 >contig.log./ SOAPdenovo map -s lib23.cfg -g ant >map.log./ SOAPdenovo scaff -g ant -F >scaff.log2 参数说明用法:/PathToProgram/SOAPdenovo all -s configFile [-K kmer -d KmerFreqCutOff -D EdgeCovCutoff -M mergeLevel -R -u -G gapLenDiff -L minContigLen -p n_cpu] -o Output-s STR 配置文件-o STR 输出文件的文件名前缀-g STR 输入文件的文件名前缀-K INT 输入的K-mer值大小,默认值23,取值范围 13-63 -p INT 程序运行时设定的线程数,默认值8-R 利用read鉴别短的重复序列,默认值不进行此操作-d INT 去除频数不大于该值的k-mer,默认值为0-D INT 去除频数不大于该值的由k-mer连接的边,默认值为1,即该边上每个点的频数都小于等于1时才去除-M INT 连接contig时合并相似序列的等级,默认值为1,最大值3。