参考westernblotting
- 格式:doc
- 大小:134.01 KB
- 文档页数:22
Western blot
原理:
通过电泳区分不同的组分,并转移至固相支持物,通过特异性试剂(抗体)作为探针,对靶物质进行检测,蛋白质的Western印迹技术结合了凝胶电泳的高分辨率和固相免疫测定的特异敏感等多种特点,可检测到低至1~5ng(最低可到10-100pg)中等大小的靶蛋白。
一、抗原的选择和制备
A:样品的制备
1 组织:
组织的处理方法:组织洗涤后加入3倍体积预冷的PBS,0℃研磨,加入5×STOP buffer,180W,6mins,0℃超声波破碎,5000rpm,5mins 离心,取上清。加入β-ME(9.5ml加入0.5ml),溴酚蓝(9.5ml加入0.5ml)煮沸10min,分装后于-20℃保存,用时取出,直接溶解上样。
2 细胞:
细胞的处理方法: 离心收集细胞或者直接往细胞培养瓶内加入5×STOP buffer,收集,180W,6mins,0℃超声波破碎,5000rpm,5mins 离心,取上清。加入β-ME(9.5ml加入0.5ml),溴酚蓝(9.5ml加入0.5ml)煮沸10min,分装后于-20℃保存,用时取出,直接溶解上样。
3 分泌蛋白的提取(特例):
直接收集分泌液,加入β-ME、溴酚蓝制样。
B:蛋白的定量方法及影响蛋白定量原因
1.双缩脲法:
双缩脲法是第一个用比色法测定蛋白质浓度的方法。在需要快速,但不很准确的测定中,常用此法。硫铵不干扰显色,这对蛋白质提纯的早期阶段是非常有利的。双缩脲法的原理是Cu2+与蛋白质的肽键,以及酪氨酸残基络合,形成紫蓝色络合物,此物在540nm波长处有最大吸收。双缩脲法常用于0.5g/L~10g/L含量的蛋白质溶液测定。干扰物有硫醇,以及具有肽性质缓冲液,如Tris、Good缓冲液等。可用沉淀法除去干扰物,即用等体积10%冷的三氯醋酸沉淀蛋白质,然后弃去上清液,再用已知体积的1m NaOH溶解沉淀的蛋白质进行定量测定。
2.Lowry法:
此法是双缩脲法的进一步发展。他的第一步就是双缩脲反应,即Cu++与蛋白质在碱性溶液中形成络合物,然后这个络合物还原磷钼磷-磷钨酸试剂(福林-酚试剂),结果得到深蓝色物。此法比双缩脲法灵敏得多,适合于测定20mg/L~400mg/L含量的蛋白质溶液。其干扰物质与双缩脲法相同,而且受他们的影响更大,硫醇和许多其他物质的存在会使结果严重偏差。另外要注意的是,加入福林试剂时要特别小心,试剂只在酸性pH环境中才稳定,上述提到的还原反应只有在pH10时才发生,因此,福林试剂加入到碱性的Cu2+-蛋白质溶液中时,必须立刻搅拌,以使磷钼酸-磷钨酸试剂在未被破坏之前能有效地被Cu2+-蛋白质络合物所还原。
3.紫外吸收法:
大多数蛋白质在280nm波长处有特征的最大吸收,这是由于蛋白质中有酪氨酸,色氨酸和苯丙氨酸存在的缘故,因此,利用这个特异性吸收,可以计算蛋白质的含量。如果没有干扰物质的存在,在280nm处的吸收可用于测定0.1~0.5mg/ml含量的蛋白质溶液。部分纯化的蛋白质常含有核酸,核酸在260nm波长处有最大吸收。
4. Bradford比色法:
Bradford比色法比Lowry法测定蛋白浓度更简单迅速。用脱氧胆酸/三氯乙酸沉淀蛋白可排除甘油、去污剂、2-巯基乙醇、乙酸、硫酸铵、Tris和一些碱性缓冲系统的干扰。
分别在两组微量离心管中各加入0.5mg/ml牛血清白蛋白(5,10,15和20祃),以0.15mmol/l
NaCl补足至100祃,同时以两管100祃的0.15mmol/l NaCl作空白对照。每管各加入1ml考马斯亮蓝染料溶液,振荡混匀,室温放置2分钟。用1cm光径的微量比色杯测A595,取A595吸光值对标准蛋白浓度作图,画标准曲线,并测量待测样品的A595。从BSA标准曲线中确定待测样品的浓度。测定10-100礸的蛋白质,要在较大试管中将染料溶液体积增大5倍进行。样品浓度过高,可稀释后进行,或在10-100礸另作一标准曲线进行测定。
5.电泳估算法(我们选择此法):
样品倍比稀释,SDS-PAGE电泳,同时做定量marker对照,可以估算样品大概浓度。
以提取癌组织总蛋白为例:
① 取等量胰腺癌组织、癌旁组织及正常胰腺组织,用dH2O漂洗5~10次,再用预冷的1×PBS洗涤3次,目的是去除样本中的血液。
② 每2克组织加入3ml 1×PBS匀浆,保持在4℃条件进行。
③ 加入5×STOP Buffer缓冲液1ml,混匀,4℃下超声碎化。再加入0.5ml β-ME,0.5ml溴酚蓝,煮沸10mins,至此,制样过程完成;
④ SDS-PAGE电泳,以BAS作为对照估计样品蛋白浓度。
二、SDS-聚丙烯酸胺凝胶电泳(SDS-PAGE)
A:实际操作
1. 做胶前的准备
1)检查是否有足够的、干净的 spacer、comb 和架子。
2)检查是否有新鲜的,足量10%APS,没有立刻重配。
3)按将要检测的抗体对应的原始抗原的分子量大小,计算出胶的浓度,并算出分离胶各组分的用量。
2. 制胶,电泳
1)装好架子。
2)按照下面配方配制分离胶。(单位:ml,Total: 8ml)
7.5% 10% 15%
2×Sep. buffer 4 4 4
30% Gel.sol 2.0 2.7 4
ddH2O 1.9 1.2 0
TEMED 8ul 8ul 8ul
10%APS 80ul 0ul 80ul
在胶上面加入一层蒸馏水,促进胶更好的凝集。
3)待分离胶凝集后,配制浓缩胶。(单位:ml,Total: 3.5ml)
3%
2×Stacking. buffer 1.7
30% Gel.sol 0.35
ddH2O 1.4
TEMED 5ul
10%APS 50ul
倒好后插入预先准备好的梳子。
4)待胶凝集好后,上样,电泳。 上层胶用60-80V电压,当样品至分离胶时,用100-120V电压。一般电泳时间在1.5小时左右。
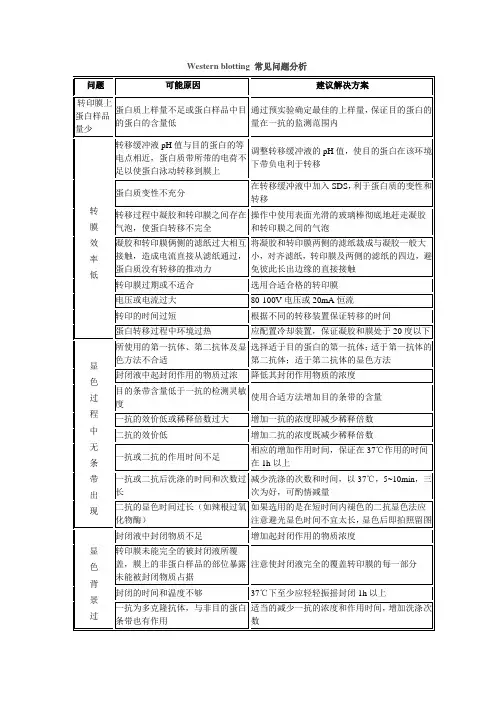
B :注意事项及常遇到的问题 1)分离胶不要倒的太满,需要有一定的浓缩胶空间,否则起不到浓缩效果。
2)上样蛋白量不应超过30ug/mm2 (载荷面即:如果你的胶槽是5mm×1mm,则载荷面为:1mm×5mm=5mm2) 。
3)gel通常在0.5-1h内凝集最好,过快表示TEMED、APS用量过多,此时胶太硬易龟裂,而且电泳时容易烧胶。太慢则说明两种试剂用量不够或者系统实际不纯或实效。
4)混合搅拌速度太快产生气泡影响聚合,导致电泳带畸形。太慢不均匀,特别是甘油。
5)电泳中常出现的一些现象:
︶ 条带呈笑脸状,原因:凝胶不均匀冷却,中间冷却不好。
︵ 条带呈皱眉状,可能是由于装置不合适,特别可能是凝胶和玻璃挡板底部有气泡,或者两边聚合不完全。
拖尾:样品溶解不好。
纹理(纵向条纹):样品中含有不溶性颗粒。
条带偏斜:电极不平衡或者加样位置偏斜。
条带两边扩散:加样量过多。
三、转移
在电流的作用下,使蛋白质从胶转移至固相载体(膜)上。
膜的选择:印迹中常用的固相材料有NC膜、DBM、DDT、尼龙膜、PVDF等。
我们选用PVDF(聚偏二氟乙烯),其具有更好的蛋白吸附、物理强度,以及具有更好的化学兼容性。有两种规格:Immobilon-P(0.45um)和Immobilon-PSQ(0.2um for MW<20kDa)。
A 半干法
即将凝胶夹层组合放在吸有转印缓冲液的滤纸之间,通过滤纸上吸附的缓冲液传导电流,起到转移的效果。因为电流直接作用在膜胶上,所以其转移条件比较严酷,但是其转移时间短,效率高。
1 实验条件的选择
电流1mA-2mA/cm2,我们通常100mA/膜,按照目的蛋白分子大小、胶浓度选择转移时间,具体可以根据实际适当调整。
目的蛋白分子大小(kDa) 胶浓度 转移时间
80---140 8% 1.5-2.0
25---80 10 1.5
15--40 12 0.75
<20 15 0.5
2 实验操作
(1)滤纸和膜的准备 (在电泳结束前20分钟应开始准备工作)。
A. 检查是否有足够的transfer buffer,没有立即配制。
B. 检查是否有合适大小的滤纸和膜。
C. 将膜泡入甲醇中,约1-2分钟。再转入transfer buffer中。
D. 将合适的靠胶滤纸和靠膜滤纸分别泡入transfer buffer 中。
PDVF膜在进转移缓冲液时,要在甲醇里面泡多长时间?
PVDF是疏水性的,在转膜缓冲液里很难浸透,甲醇处理后使更容易浸润。 PVDF的预处理是用甲醇浸泡活化,浸泡透之后转入蒸馏水洗两次。用甲醇泡的目的是为了活化PVDF膜上面的正电基团,使它更容易跟带负电的蛋白质结合。
一般5-20分钟都有人在用。可在取胶的同时泡,估计前后5分钟左右。效果还不错。以前有站友发贴,说处理时间没多大关系,关键看膜的质量.确实是这样的。只要让膜完全浸透应该就没问题了。
Hybond的PVDF说明书这样的写的:“pre-wet the membrane in 100% methanol (10 seconds)”。
我的理解是,至少10秒钟,多泡下也没关系的,事实上,泡10秒的做过,10分钟的也做过,都没有问题的。
(2)转移
A. 在电转移仪上铺好下层滤纸。一般用三层。
B. 将膜铺在靠膜滤纸上,注意和滤纸间不要有气泡,再倒一些transfer buffer到膜上,保持膜的湿润。
C. 将胶剥出,去掉stack gel,小心的移到膜上。
D. 剪去膜的左上角,在膜上用铅笔标记出胶的位置。
E. 将一张靠胶滤纸覆盖在胶上。 倒上些transfer buffer,再铺两张靠胶滤纸。
F. 装好电转移仪,根据需要选定所需的电流和时间。
G. 转移过程中要随时观察电压的变化,如有异常应及时调整。
3 注意事项及常遇到的问题
1)滤纸、胶、膜之间的大小,一般是 滤纸>=膜>=胶?
2)滤纸、胶、膜之间千万不能有气泡,气泡会造成短路。
3)因为膜的疏水性,膜必须首先在甲醇中完全浸湿。而且在以后的操作中,膜也必须随时保持湿润(干膜法除外)。
4)滤纸可以重复利用,上层滤纸(靠膜)内吸附有很多转移透过的蛋白质,所以上下滤纸一定不能弄混,在不能分辨的情况下,可以将靠胶滤纸换新的。
5)转移时间一般为1.5 小时,( 1mA-2mA/cm2,10% gel),可根据分子量大小调整转移时间和电流大小。
1)在叠膜、胶、纸三明治时候,操作于盛有转膜Buffer的大平皿中进行,叠好后,再将三者平行转移至转膜装置,切勿再移动,因为在buffer中操作,已经完全排除了气泡存在的可能,无需再赶气泡
2)关于转膜时间:半干转的话,20 V×30min即可
B 湿法
我们基本上不用此方法,这里暂不做介绍。
C 转移后效果的鉴定
1.染胶
用考马斯亮兰染色经destain脱色后,看胶上是否还有蛋白来反映转移的效果。