多重序列比对
- 格式:ppt
- 大小:3.43 MB
- 文档页数:1
第十章 多重序列比對 Vector NTI的多重序列比對程式和其他的比對軟體比較起來非常的方便實用,操作介面也很簡單,比對的結果可以存取和輸出。
NTI有兩種序列比對程式,一種為AlignX,可以用在核酸序列和蛋白質序列比對;另一種為AlignX Blocks,只能用在蛋白質序列比對。
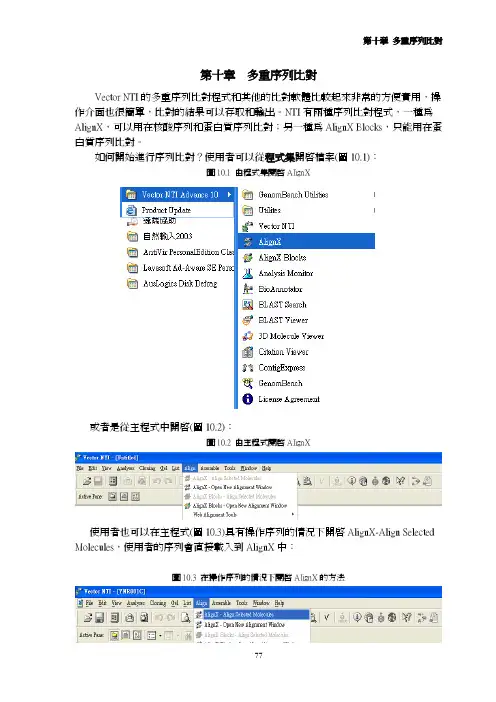
如何開始進行序列比對?使用者可以從程式集開啟檔案(圖10.1):圖10.1 由程式集開啟AlignX 或者是從主程式中開啟(圖10.2):圖10.2 由主程式開啟AlignX 使用者也可以在主程式(圖10.3)具有操作序列的情況下開啟AlignX-Align Selected Molecules,使用者的序列會直接載入到AlignX中:圖10.3 在操作序列的情況下開啟AlignX的方法 開啟AlignX之後,使用者會見到圖10.4的畫面:圖10.4在操作序列的情況下開啟AlignX首先使用者要把序列載入Vector NTI程式中,可以點選或者從左上方的Project→Add Files把序列檔案載入,請注意檔案名不可以過長,檔名過長會造成程式進行比對時無法完全顯示檔名(圖10.5):圖10.5 輸入的檔名注意不可過長 選取檔案後按下開啟就可以載入程式中,若比對的序列很多時可以用滑鼠圈選欲分析的序列後選擇開啟。
序列檔案載入的時候程式會詢問該序列為核酸序列或是蛋白質序列,點選好以後再點選Import就可以了(圖10.6):圖10.6 載入時,會詢問序列的性質,核酸序列或蛋白質序列接下來程式的左上方會出現使用者載入的序列(圖10.7),序列載入完成以後就可以開始進行比對的操作:圖10.7 成功載入序列的畫面進行比對前,先把欲比對的序列用滑鼠進行圈選(圖10.8):圖10.8 選取欲比對之序列只要按下或是從上方Align→Align Selected Sequence(圖10.9)就會進行比對運算:圖10.9按下Align→Align Selected Sequence進行比對運算好以後就會出現下面的畫面(圖10.10);圖10.10 比對完的結果 分析完成後畫面(圖10.11)會出現比對的相關結果,最下方是序列比對的圖形,左邊中間的區塊所顯示的圖形為導引樹(Guide tree),用來表示序列之間的關連性。

生物信息学中的多重序列比对算法生物信息学是一门交叉学科,主要研究生物体内的相关信息,如基因、蛋白质等,与计算机科学相结合,开发相应的算法和软件来处理这些信息。
多重序列比对是生物信息学中一个基本的、重要的问题,在基因组学和系统生物学研究中有着广泛的应用。
本文将会介绍多重序列比对的背景和意义,并着重讨论多种常见的多重序列比对算法。
一、多重序列比对的背景和意义DNA序列中的每一个碱基都是遵循特定的规律排列而成的,对于同一物种不同个体的DNA序列中,虽然具有相同的碱基种类,但在具体的分布和数量上,还是会存在一定的差异。
这些差异可能涉及到基因的表达、蛋白质的功能以及遗传变异等方面。
因此,通过对多个DNA序列进行比对,可以发现它们之间的差异和联系,从而深入了解物种的演化路径和生物功能等方面。
多重序列比对的具体过程是将多条序列进行比对,找出它们之间的共同区域和不同之处。
而这个过程并不是一件轻松的事情,因为序列长度的不同和存在的错配等现象,这个比对过程难点很多。
因此,多重序列比对算法的研究和发展也成为了生物信息学研究的前沿领域之一。
二、多重序列比对算法概述多重序列比对算法根据方法不同,可以分为两类,一种是基于全局比对的算法,另一种则是基于局部比对的算法。
在全局比对中,整条序列被视为一个整体进行比对;而在局部比对中,仅比对序列中的一部分区域,这个区域通常是各个序列中比较相似的地方。
下面分别介绍几个常见的多重序列比对算法:1. ClustalWClustalW是一种全局比对算法,它是一种基于序列之间的距离矩阵进行序列比对的方法。
在ClustalW中,首先将多个序列之间的距离计算出来,然后根据距离矩阵的结果进行多序列比对。
ClustalW算法具有速度快、易于使用的特点。
但是,它的精确度不高,适合处理比较简单的序列之间的比对。
2. MuscleMuscle是一种全局比对算法,其特点是能够使用多种方法来计算序列之间的距离矩阵,常见的包括kmer覆盖率、Poisson模型等。

题目A :多重序列比对的数学模型与算法自美国提出组织的人类基因组计划(Human Genome Proreet )简称为HGP 以来,美国每年拔出相当大的经费支持,日本、法国、英国、德国等纷纷响应,它们的工作使新的交叉学科生物信息论得以诞生和发展,生物信息论是用数理和信息科学的观点、理论和方法去研究生命现象,组织和分析呈指数增长的生物学数据。
生物信息学是一门综合学科,是计算机科学、数学、物理、生物学的结合。
生物信息学的基础是各种数据库的建立和分析工具的发展。
目前,生物学数据库已达500个以上,共有四大类:基因组数据库,核酸和蛋白质一级结构数据库、生物大分子三维空间结构数据库及其以她们为基础构建的二级数据库。
生物信息学主要研究基因组测序及其信息分析、生物大分子的结构与功能预测及其模拟和药物设计、大规模基因表达数据的分析与基因芯片设计,以及基因与蛋白质相互作用网络等四方面的问题。
多重序列比对是计算分子生物学中最重要的运算。
多重序列比对的基本问题就是找出适当安排删减与插入尽量少的空格,使得两个序列达到最大程度的一致的方案。
比如给出下列三个序列:_ (1)AC GAGTCC ACT我们适当安排删减与插入空格得到:_____ (2)___ACG A GTCC AC T(2)就是多重序列的一个比对。
局部分段比对是其中更为常见的运算。
上世纪80年代,Smith-Waterman 提出了两个序列的局部比对的明确的模型。
1998—1999年,相继出现利用k-tuple 的快速容错分段比对搜索法。
2002年开始出现对完整基因组及其异常基因的比较研究以及多重序列比对问题的研究,2003年刘军Mayetri Gupta 和刘军得到Motif 的搜索算法。
人类基因组计划后,目前已经进入后基因时代,主要就是对人类基因组计划实施得到的基本数据库进行信息分析、加工和利用,提取有用信息,用来研究生命现象中的重大问题。
多重序列比对问题是生物信息学的基本问题,多重序列比对技术也是生物信息学的基本工具,有着十分广泛的应用,比如基因是否为同一个家族,癌症患者的基因与正常时的基因比对分析等等。

第十章多重序列比对 Vector NTI的多重序列比对程序和其他的比对软件比较起来非常的方便实用,操作接口也很简单,比对的结果可以存取和输出。
NTI有两种序列比对程序,一种为AlignX,可以用在核酸序列和蛋白质序列比对;另一种为AlignX Blocks,只能用在蛋白质序列比对。
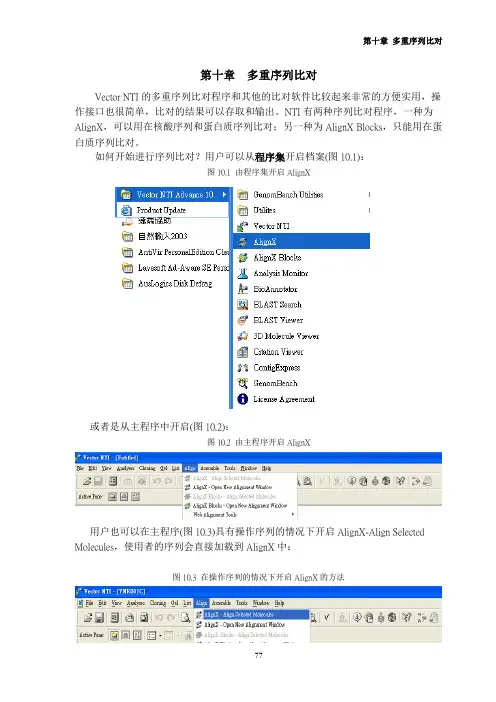
如何开始进行序列比对?用户可以从程序集开启档案(图10.1):图10.1 由程序集开启AlignX 或者是从主程序中开启(图10.2):图10.2 由主程序开启AlignX 用户也可以在主程序(图10.3)具有操作序列的情况下开启AlignX-Align Selected Molecules,使用者的序列会直接加载到AlignX中:图10.3 在操作序列的情况下开启AlignX的方法 开启AlignX之后,使用者会见到图10.4的画面:图10.4在操作序列的情况下开启AlignX首先用户要把序列加载Vector NTI程序中,可以点选或者从左上方的Project →Add Files把序列档案加载,请注意文件名不可以过长,檔名过长会造成程序进行比对时无法完全显示文件名(图10.5):图10.5 输入的档名注意不可过长 选取档案后按下开启就可以加载程序中,若比对的序列很多时可以用鼠标圈选欲分析的序列后选择开启。
序列档案加载的时候程序会询问该序列为核酸序列或是蛋白质序列,点选好以后再点选Import就可以了(图10.6):图10.6 载入时,会询问序列的性质,核酸序列或蛋白质序列接下来程序的左上方会出现使用者加载的序列(图10.7),序列加载完成以后就可以开始进行比对的操作:图10.7 成功载入序列的画面进行比对前,先把欲比对的序列用鼠标进行圈选(图10.8):图10.8 选取欲比对之序列只要按下或是从上方Align→Align Selected Sequence(图10.9)就会进行比对运算:图10.9按下Align→Align Selected Sequence进行比对运算好以后就会出现下面的画面(图10.10);图10.10 比对完的结果 分析完成后画面(图10.11)会出现比对的相关结果,最下方是序列比对的图形,左边中间的区块所显示的图形为导引树(Guide tree),用来表示序列之间的关连性。

实验四基于CLUSTAL算法的多重序列比对分析1. CLUSTAL简介CLUSTAL是对核苷酸或蛋白质进行多序列比对的程序,也可以对来自不同物种的功能相同或相似的序列进行比对和聚类,通过构建系统发生树判断亲缘关系,并对序列在生物进化过程中的保守性进行估计。
CLUSTAL有CLUSTALX和CLUSTALW之分,CLUSTALW 是以命令行格式运行,CLUSTALX则通过窗口格式进行操作。
目前最新版本为CLUSTAL 1.83,均可以从ftp:///pub/software/下载。
这里我们主要介绍CLUSTAL W,从ftp直接下载DOS文件夹下的CLUSTAL W到本地磁盘解压,其中有两个exe文件,CLUSTALW.exe是进行多序列比对和生成亲缘树的程序,而njplotWIN95则是对CLUSTALW.exe运行结果进行察看的程序。
另外还有许多在线的Clustal W服务,例如:/Clustalw/2 . 本地运行Clustal WClustal W程序能自动识别输入的序列,通常当读入的序列字母85%以上为A、C、G、T、U或N时,则被认为是核苷酸序列,反之为蛋白质序列。
进行多序列比对时,要求所有输入的序列按顺序储存于一个文件中。
当有大量的序列文件时,可以在Unix操作系统下用cat file1.seqfile2.seq……>multiseq.seq命令合并成一个文件序列的储存格式必须为以下7种格式之一,他们分别是:NBRF/PRI、EMBL/SWISSPORT、Pearson(Fasta)、Clustal(*.aln)、GCG/MSF(Pileup)、GCG9/RSF和GDE,除了“-”和“.”外所有的非字母都将被忽略。
这里我们将不同来源的15条甲硫酰胺tRNA 合成酶的氨基酸序列,保存在单一文件multiseq.file中。
进入程序安装目录,双击CLUSTALW.exe文件,进入Clustal W的主菜单界面(见图1)。

基于多重序列比对的蛋白质结构预测研究蛋白质是生命体内最为重要的分子之一,其结构决定了其功能。
因此,蛋白质结构预测是生物信息学领域的一个重要研究方向。
在蛋白质结构预测中,多重序列比对是一种常用的方法,因其能够从多个相关蛋白序列中提取信息以预测蛋白质的结构。
1. 比对算法选择多重序列比对算法有多种,如MAFFT、ClustalW、T-Coffee等。
这些算法基于不同的策略和算法设计,各自有其优缺点。
在选择算法时需要依据比对的目的和序列的性质进行考虑。
2. 数据库建设多重序列比对需要大量的序列数据支持,因此需要建设一套完整的蛋白质序列数据库。
该数据库应该包含已知的蛋白质序列以及未知的序列,以便预测新蛋白质的结构。
此外,还需要对序列进行预处理,如去除无效的序列、标准化序列等。
3. 特征提取在多重序列比对中,还需要对序列进行特征提取。
这些特征包括氨基酸成分、结构域、残基间距等信息。
特征提取的目的是为了减少不必要的噪音干扰,并提高模型的准确性。
4. 结构预测在完成多重序列比对和特征提取后,就可以开始进行蛋白质结构预测。
最常见的方法是利用机器学习算法,如支持向量机、神经网络、深度学习等,对序列特征进行建模。
这些模型可以在未知序列中预测蛋白质的结构。
5. 优化方法目前,在多重序列比对中,依然存在一些挑战和限制。
其中最主要的问题是维度灾难和过拟合。
因此,在优化方法方面,可以采用降维技术、交叉验证等方法来解决这些问题。
综上所述,基于多重序列比对的蛋白质结构预测是一个具有挑战性的领域。
在今后的研究中,需要不断深入理解相关算法和技术,加强对蛋白质结构的理解,提高预测的准确性,为生物学界研究更多复杂生命体提供更好的方案。

C l u s t a l x多重序列比对图解教程(B y R a i n d y) 本帖首发于Raindy'blog软件简介:CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
ClustalX为进行多重序列和轮廓比对和分析结果提供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征。
窗口上面的下拉菜单可让你选择传统多重比对和轮廓比对需要的所有选项。
主要功能:你可以剪切、粘贴序列以更改比对的顺序;你可以选择序列子集进行比对;你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx1.81版链接地址:ist&ID=7435(请完整复制)应用:Clustalx比对结果是构建系统发育树的前提实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方的菜单栏选择“File”-“LoadSequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是否替换现有序列(Replaceexistingsequences),根据具体情形选择操作。
图1图22.编辑序列:对标尺(Ruler)上方的序列进行编辑操作,主要有Cutsequences(剪切序列)、Pastesequences(粘贴)、SelectAllsequences(选定所有序列),ClearsequenceSelection(清除序列选定)、Searchforstring(搜索字串)、RemoveAllgaps(移除序列空位)、RemoveGap-OnlyColumns(仅移除选定序列的空位)图33.参数设置:可以根据分析要求设置相对的比对参数。
序列比对的理论基础是进化学说:如果两个序列之间具有足够的相似性,就推测二者可能有共同的进化祖先,经过序列内残基的替换、残基或序列片段的缺失、以及序列重组等遗传变异过程分别演化而来。
序列相似和序列同源是不同的概念,序列之间的相似程度是可以量化的参数,而序列是否同源需要有进化事实的验证。
物以类聚人以群分,就像你要了解一个人可以通过了解他的朋友一样,序列比对是从已知获得未知的一个十分有用的方法。
另外,物种亲缘树的构建都需要进行生物分子序列的相似性比较。
序列比对按照数目、范围和对象来分,可以分为:o两序列比对和多序列比对o全局比对和局部比对o核酸序列比对和氨基酸序列比对。
限于篇幅,今天只给大家介绍如何使用DNAMAN 8作核酸多序列比对。
多序列比对就是把两条以上可能有系统进化关系的序列进行比对的方法。
其意义在于它能够把不同种属的相关序列的比对结果按照特定的格式输出,并且在一定程度上反映它们之间的相似性。
首先,在解螺旋回复0628下载DNAMAN 8软件。
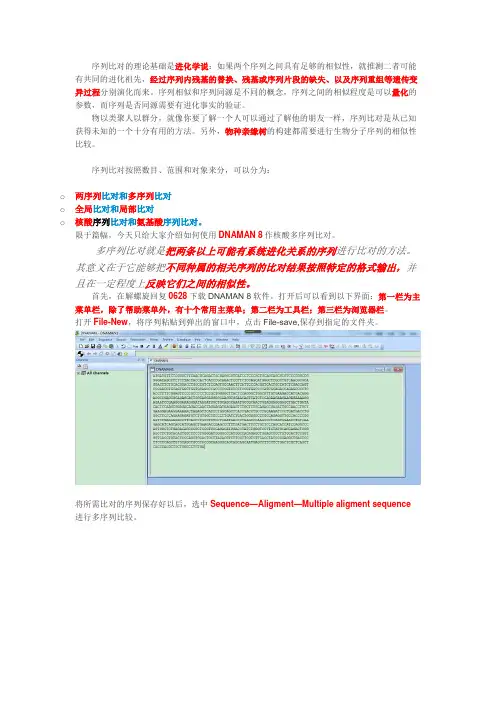
打开后可以看到以下界面:第一栏为主菜单栏,除了帮助菜单外,有十个常用主菜单;第二栏为工具栏;第三栏为浏览器栏。
打开File-New,将序列粘贴到弹出的窗口中,点击File-save,保存到指定的文件夹。
将所需比对的序列保存好以后,选中Sequence—Aligment—Multiple aligment sequence 进行多序列比较。
在弹出的窗口Sequence&Files中加载序列,File、Fold、channel、Database分别表示从文件、文件夹、channel和数据库中获取序列。
勾选窗口中的“DNA”,点击“下一步”。
在弹出的窗口Method中,“optimalaligment”最佳比对方式中有四个高大上的选项:Full Alignment(完全比对)、Prosile Aligment(轮廓比对)、New Swquence on Profile (轮廓上的新序列)、Fast Alignment(快速比对),本文选择了Fast Alignment,并且勾选了Try both strands(尝试使用双链)。

生物信息学中多重序列比对算法研究多重序列比对是生物信息学领域中的一个重要任务,它用于对多个生物序列进行比较和分析,从而揭示它们之间的共同点和差异。
多重序列比对广泛应用于基因组学、进化生物学和药物研发等领域,对于理解基因和蛋白质序列的功能和结构起着关键作用。
本文将介绍一些常见的多重序列比对算法及其应用。
1. 概述多重序列比对是通过将多个生物序列进行配对,找出相同和相似的区域以及揭示序列差异的一种方法。
它可以帮助研究人员理解进化相关的序列保守性、功能域、结构域和突变位点等信息。
多重序列比对算法的主要挑战在于在保证准确性和效率的前提下,应对序列长度和数量的增加所引起的计算复杂性增加。
2. 算法分类目前,多重序列比对算法可以分为两大类:多序列动态规划方法和高效启发式方法。
2.1 多序列动态规划方法多序列动态规划方法将多重序列比对问题转化为在一个多维矩阵中求解最优路径。
其中最著名的算法是Progressive MSA(渐进性多重序列比对算法)。
该算法以两两序列比对为基础,在不同的聚类层次上逐步合并序列,直到得到最终的多重序列比对结果。
另外一种常见的算法是POA(Partial Order Alignment),它通过构建序列树和部分序列的插入来进行多重序列比对。
2.2 高效启发式方法高效启发式方法通过使用一些策略和技巧来减少计算复杂性和提高算法效率。
其中最著名的算法是MUSCLE(Multiple Sequence Comparison by Log-Expectation),它使用迭代聚类和改进的目标函数来进行多重序列比对。
在实践中,MUSCLE通常比Progressive MSA更快并能够得到同样准确的结果。
另外一种常见的算法是MAFFT(Multiple Alignment using Fast Fourier Transform),它利用傅立叶变换的思想将多重序列比对问题转化为一个大规模矩阵相乘的问题,从而提高算法效率。
MSA:多重⽐对序列的格式及其应⽤多重⽐对序列的格式及其应⽤这⾥对多重序列⽐对格式(Multiple sequence alignment – MSA)进⾏总结。
在做系统演化分析、序列功能分析、基因预测等,都需要涉及到多重序列⽐对。
特别是当需要⽤不同软件对多重⽐对序列进⾏批量操作时,会遇到各种的格式,⽽这些格式是如何产⽣的,有什么区别,格式之间如何转换,从哪⾥可以下载到相关的格式序列,不同的格式⼜有什么特殊的⽤途等,本篇⽂章将就这些问题进⾏总结与讨论。
因为涉及内容较多,不⾜之处,欢迎⼤家补充或者批判。
⽣物信息学的基础是基于这样的⼀个假设:序列相似,结构相似,功能相似。
所以相似的⼀组序列,就可能同属于⼀个基因家族,⽽这样的⼀组序列相似的部分,就可能使其功能之所在,称其为结构域。
这是对于基因家族分类的⼀种⽅式,将结构与功能进⾏联系,从⽽实现从结构预测功能(序列称为⼀级结构)。
进⾏多重⽐对、多重序列的编辑、多重序列注释、存储与展⽰、系统演化分析等,不同的软件、不同的系统,除了要兼容现有的格式,还会根据⾃⾝的需要,都定义新的格式。
所以这些本⾝可以进⾏部分的格式转换,同时许多脚本模块⽐如bioperl等也提供了⼀些格式之间转换的脚本。
这些格式同发布其软件平台有着密切的联系,随着软件的流⾏⽽流⾏。
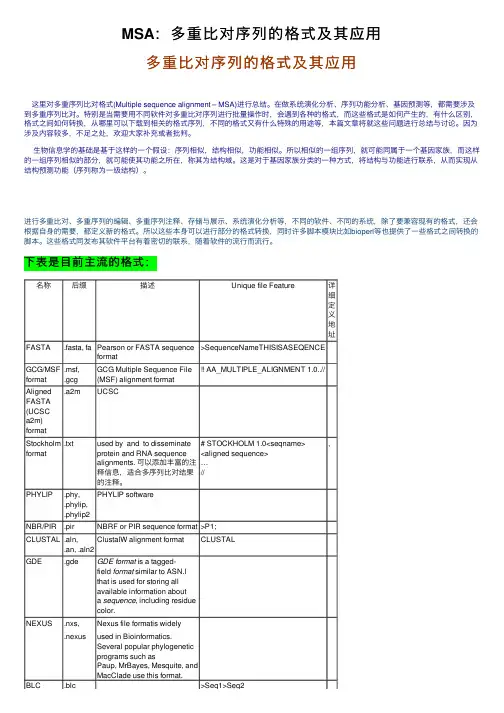
下表是⽬前主流的格式:名称后缀描述Unique file Feature详细定义地址FASTA.fasta, fa Pearson or FASTA sequenceformat>SequenceNameTHISISASEQENCEGCG/MSF format .msf,.gcgGCG Multiple Sequence File(MSF) alignment format!! AA_MULTIPLE_ALIGNMENT 1.0..//AlignedFASTA(UCSCa2m)format.a2m UCSCStockholm format .txt used by and to disseminateprotein and RNA sequencealignments. 可以添加丰富的注释信息,适合多序列⽐对结果的注释。
多序列比对及系统进化树的构建【实验目的】1、掌握使用Clustalx进行序列多重比对的操作方法;2、熟悉构建分子系统发生树的基本过程,掌握使用相关软件构建系统发生树的操作方法。
【实验原理】在现代分子进化研究中,根据现有生物基因或物种多样性来重建生物的进化史是一个非常重要的问题。
一个可靠的系统发生的推断,将揭示出有关生物进化过程的顺序,有助于了解生物进化的历史和进化机制。
对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行比对。
⑵要构建一个进化树(phyligenetic tree)。
⑶对进化树进行评估,主要采用Bootstrap法。
进化树的构建是一个统计学问题,所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。
模拟的进化树需要一种数学方法来对其进行评估。
CLUSTALX和MEGA软件能够实现上述的建树步骤。
CLUSTALX是Windows界面下的多重序列比对软件。
MEGA是多个软件的压缩包,功能极其强大,主要包括五个方面的功能软件:i,DNA和蛋白质序列数据的分析软件。
ii,序列数据转变成距离数据后,对距离数据分析的软件。
iii,对基因频率和连续的元素分析的软件。
iv,把序列的每个碱基/氨基酸独立看待(碱基/氨基酸只有0和1的状态)时,对序列进行分析的软件。
v,按照DOLLO简约性算法对序列进行分析的软件。
vi,绘制和修改进化树的软件。
【实验内容】1、使用CLUSTALX软件对一组蛋白质序列(leptin.txt)进行多重序列比对;2、使用MEGA 软件包构建上述DNA分子系统发生树。
【实验方法】一、用CLUSTALX软件对已知序列做多序列比对。
1、在NCBI数据库搜索人leptin的同源蛋白序列2、下载leptin的同源蛋白序列8-10条,以FASTA格式保存为leptin.txt文件。
2、双击进入CLUSTALX程序,点FILE进入LOAD SEQUENCE,打开leptin.txt文件。
第三章序列比较3.3 序列多重比对与序列两两比对不一样,序列多重比对(Multiple Alignment)的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系和推测它们的结构、功能,那么,同时比对一组序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似性只能通过将多个序列对比排列起来才能识别。
同样,只有在多序列比对之后,才能发现与结构域或功能相关的保守序列片段。
对于一系列同源蛋白质,人们希望研究隐含在蛋白质序列中的系统发育的关系,以便更好地理解这些蛋白质的进化。
在实际研究中,生物学家并不是仅仅分析单个蛋白质,而是更着重于研究蛋白质之间的关系,研究一个家族中的相关蛋白质,研究相关蛋白质序列中的保守区域,进而分析蛋白质的结构和功能。
序列两两比对往往不能满足这样的需要,难以发现多个序列的共性,必须同时比对多条同源序列。
图3.14是从多条免疫球蛋白序列中提取的8个片段的多重比对。
这8个片段的多重比对揭示了保守的残基(一个是来自于二硫桥的半胱氨酸,另一个是色氨酸)、保守区域(特别是前4个片段末端的Q-PG)和其他更复杂的模式,如1位和3位的疏水残基。
实际上,多重序列比对在蛋白质结构的预测中非常有用。
多重比对也能用来推测各个序列的进化历史。
从图3.14可以看出,前4条序列与后4条序列可能是从两个不同祖先演化而来,而这两个祖先又是由一个最原始的祖先演化得到。
实际上,其中的4个片段是从免疫球蛋白的可变区域取出的,而另4个片段则从免疫球蛋白的恒定区域取出。
当然,如果要详细研究进化关系,还必须取更长的序列进行比对分析。
对于多重序列比对的定义,实际上是两个序列的推广。
设有k个序列s1, s2, ... ,s k,每个序列由同一个字母表中的字符组成,k大于2;通过插入操作,使得各序列s1, s2, ... ,s k的长度一样,从而形成这些序列的多重比对。
如果将各序列在垂直方向排列起来,则可以根据每一列观察各序列中字符的对应关系,如图3.14。
氨基酸序列多重序列比对颜色
【絮语】
多重比对是核苷酸和氨基酸序列分析的重要内容之一,通过多重比对,可以找出序列的保守区域,可以为分子进化分析提供依据。
但由于软件的功能限制,一些比对软件生成的比对文件,如Clustalx 的*.aln文件,可读性差,无法满足高质量期刊的要求。
因此,在实际操作过程中,需要对一些多重序列比对的文件进行着色美化。
【类别】
着色美化依据划分标准不同,有多种着色类型。
(1)根据着色操作的智能程度,可分为自动着色模式和手动着色模式,如:SeqLogo和GeneDoc;
(2)根据着色色彩效果不同,则可以分为黑白模式和彩色模式,如:Boxshade和ESpript;
(3)根据联网方式,则可以分为在线方式和本地(离线)方式,如:Boxshade和GeneDoc;
(1)Boxshade 着色美化(仅黑白模式,在线运行)。
MAFFT多重序列⽐对--(附⽐对彩标⽅法)[转记]MAFFT多重序列⽐对图解教程【絮语】 ⼀提到多重序列⽐对,很多⼈禁不住就想到ClustalW(Clustalx为ClustalW的GUI版),其实有⼀款多重序列⽐对软件-MAFFT,不论从⽐对速度(Muscle>MAFFT>ClustalW>T-Coffee),还是⽐对准确性(MAFFT>Muscle>T-Coffee>ClustalW)来说,其相⽐于ClustalW(或ClustalX)有过之⽽⽆不及,所以这⾥强烈推荐使⽤MAFFT这款多重⽐对软件。
PS: 不同⽐对软件的⽐较,有兴趣的童鞋可以下载这篇⽂章看看:Alignment uncertainty and genomic analysis. Science, 2008 MAFFT官⽅⽹站: ⽀持平台:Mac OS X 、Linux、Windows Windows 32位版本:,64位版本:,请根据⾃⼰操作系统选择相应版本下载。
图1 MAFFT主界⾯ 简明操作流程: 1.载⼊序列⽂件 将FASTA格式的待⽐对序列⽂件(如:TMV.fas)复制MAFFT的根⽬录下(当然也可以放任意位置,只有找得到),双击“mafft.bat”启动MAFFT,此时提⽰输⼊⽂件(Input file?),在@后⾯输⼊⽰例的TMV.fas,也可以直接将⽂件拖⼊窗⼝(注意有个+,说明当前是拖放状态),如下图所⽰: 加载后回车,当显⽰“OK”时说明载⼊⽂件成功。
2.设置输出信息 输出⽂件名称⾃定义,扩展名任意,这⾥保留原扩展名,输出⽂件名为TMV-out.fas,确定后回车。
输出⽂件格式,建议⽤3或4,这⾥在@后输⼊4后回车,此时出现 MAFFT三种主要⽐对策略的5个选项,如下图: 当你⽆法确定时候,建议⽤第1种的--auto模式,让MAFFT根据序列的特点⾃动选择相应的⽐对策略,输⼊1后回车。