多序列比对工具-clustalX-生物在线
- 格式:ppt
- 大小:985.50 KB
- 文档页数:62


Clustalx的中文使用说明书
生物
用ClustalX做多序列比对分析图示
1、打开程序如下图所示:
2、Load Sequnce, 载入序列如下图所示:
3、选择序列文件,FASTA格式的如下图所示:
4、用文本编辑器察看FASTA序列文件内容,这里用的是记事本,推荐用EditPlus或者Ultraedit 如下图所示:
5、序列Load进去之后如下图所示:
6、Do Complete Alignment, 通常情况下直接选这个即可,无须修改比对参数如下图所示:
7、点Do Complete Alignment之后弹出的文件对话框,.dnd的是输出的指导树文件,.aln 的是序列比对结果,它们都是纯文本文件如下图所示:
点“ALIGN”之后开始等待,如果序列不多,很快就可以算完,如果数据很多,可能要等一段时间,这时候可以用眼睛盯着ClustalX的状态栏,那里会有程序运行状态和现在正在比对那两条序列的提示信息,看看可以消磨时间。
8、比对结束之后,我们可以看到这个结果如下图所示:
9、这时候我们可以发现ClustalX已经生成了.dnd和.aln两个文件,仍然用文本编辑器打开来看,这时.aln文件,这个文件可以用Mega2做进一步的bootstrap进化树分析如下图所示:
10、这是.dnd文件(指导树) 如下图所示:
11、可以用Treeview打开dnd文件,看上去就像这样子如下图所示
图3-15 ClustalX所识别的文件输入格式。

生物信息学中的序列比对工具对比总结序列比对是生物信息学中的核心技术之一,它是通过对比两个或多个生物序列的相似性和差异性来研究其结构、功能和演化关系的重要方法。
为了进行序列比对,科学家们开发了许多不同的序列比对工具。
本文将对一些常用的序列比对工具进行对比和总结。
1. BLAST (Basic Local Alignment Search Tool)BLAST 是最常用的序列比对工具之一。
它可以在短时间内快速比对大量生物序列。
BLAST 提供了多种不同的比对算法,包括常见的BLASTN(nucleotide序列比对)和BLASTP(蛋白质序列比对)。
BLAST 的优点是速度快、易用性好,适用于快速筛选大量相似序列。
2. ClustalWClustalW 是多序列比对的常用工具之一。
它使用多重序列比对算法,将多个序列的相似部分按照最佳的方式对齐。
ClustalW 可以在网页界面或命令行中使用,对于中小规模的序列比对非常高效。
3. MUSCLE (MUltiple Sequence Comparison by Log-Expectation)与ClustalW 类似,MUSCLE 也是一种常用的多序列比对工具。
它采用较新的比对算法,能够更加准确和高效地进行大规模序列比对。
MUSCLE 的优点是能处理大量序列,且能够生成高质量的比对结果。
4. MAFFT (Multiple Alignment using Fast Fourier Transform)MAFFT 是一种高性能的多序列比对工具,其算法基于快速傅立叶变换。
它可以处理大规模序列,且比对结果质量高。
MAFFT还提供了许多可选参数,以满足用户对比对过程的个性化需求。
5. T-Coffee (Tree-based Consistency Objective Function for Alignment Evaluation)T-Coffee 是一种基于树的多序列比对工具,它利用树模型来提高序列比对的准确性。

实验三、多序列比对一、软件平台clustalX、bioedit、DnaMan二、过程Clustal:○1Load Sequence(数据文件必须在ClustalX目录里)○2菜单Alignment->Alignment Parameters->Multiple Alignment Parameters 进入参数设置页面○3alignment -> do complete alignment,进行完全比对(生成.dnd和.aln 文件)○4比对完成,选择保存结果文件的格式phy:File->Save Sequence as-> 结果处理:Bioedit: ○1导入.aln文件○2“掐头去尾”editDnaMan: ○1打开DnaMan,依次打开“文件/打开指定的/多重比对”,载入Clustal X比对后的.aln文件○2点击options,参数设置,在这里,你可以设置每行显示的序列,是否显示一致序列,彩色或黑白等○3点击Output,输出为图形文件实验五、分子进化与系统发育分析一、软件平台clustalX ,MEGA,Phylip(注:phylip使用方法可搜“phylip软件的说明”)TreeView二、实验过程ClustalX:(1)使用CLUSTALX多序列比对,输出格式为*.PHY(具体见上文)(2)下载phylip,双击打开SEQBOOT ,按路径输入刚才生成的*.PHY文件;设定适当参数(4n+1);输出outfile1文件。
(3)打开PROTPARS(最大简约性法)【可选,具体情况具体分析】,输入outfile1文件后,得到outfile2和outtree1;(4)打开CONSENSE程序,输入outtree2,运行输出outfile3和outtree3文件;(5)树文件outtree3用TREEVIEW软件打开显示MEGA软件:(1)File->open a file/session->打开fasta文件,选择相应的data type (2)Align->edit/build aligns->Retrieve sequences from a file,打开文件;进行多序列比对,并另存为.meg文件(3)点击Phylogeny 选项,选择建树方法,建树保存。

Clustalx多序列比对-生物信息学实验三:多条序列比对——Clustalx实习目的:了解掌握Clustalx软件的应用,学会做多条序列比对并分析。
实习内容:一、ClustalX的使用Clustal是一种利用渐近法(progressive alignment)进行多条序列比对的软件。
即从多条序列中最相似(距离最近)的两条序列开始比对,按照各个序列在进化树上的位置,由近及远的将其它序列依次加入到最终的比对结果。
1. 准备要比对的序列请查找至少存在于5个物种中的同源序列(核酸或蛋白质皆可),并保存为fasta格式,存为文本文件(所有的序列请粘贴到同一个文本文件中)。
选择NM、XM或NP打头的序列,不要选择NC或NW打头的序列,那是全基因组序列。
建议关键词:hemoglobin,trypsin, peroxidase, p53, Superoxide Dismutase, h5n1, etc.2. 打开clustalX程序开始菜单,程序,clustalX2- clustalX23. 载入序列点最上方的File菜单,选择Load Sequence-选择你刚保存的序列文件,点打开。
”后的字符。
注意:ClustalX程序无法识别汉字,无法识别在左侧窗口里是fasta格式序列的标识号,取自序列第一行“>带空位的文件夹名,如 my document。
各位同学的序列文件不要保存在桌面上或带汉字的文件夹中,推荐保存在D盘根目录下。
4. 比对参数的选择可以对两条序列比对的参数和多条序列比对的参数进行设置。
a. 两条序列比对的参数设置点击Alilgnment菜单,选择Alignment Parameters,再选择Pairwise Alignment Parameters。
首先可以选择比对的效果,是slow/accurate 还是fast/approximate。
第一种模式采用的是动态规划算法进行比对的,第二种模式采用的是启发式的算法。
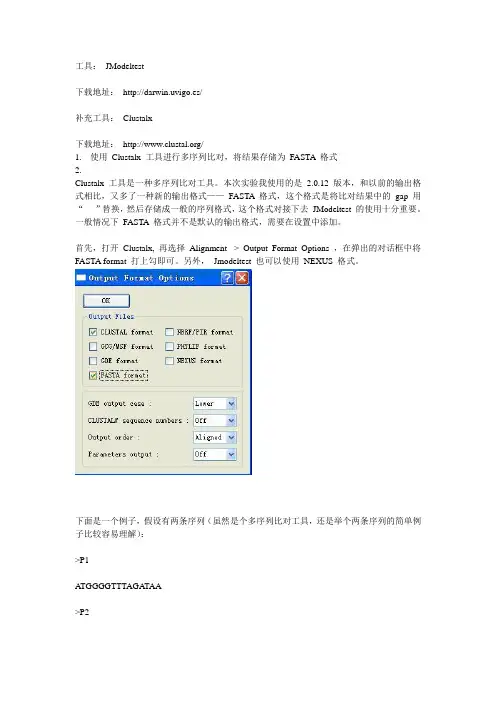
工具:JModeltest下载地址:http://darwin.uvigo.es/补充工具:Clustalx下载地址:/1.使用Clustalx 工具进行多序列比对,将结果存储为FASTA 格式2.Clustalx 工具是一种多序列比对工具。
本次实验我使用的是2.0.12 版本,和以前的输出格式相比,又多了一种新的输出格式——FASTA 格式,这个格式是将比对结果中的gap 用“- ”替换,然后存储成一般的序列格式,这个格式对接下去JModeltest 的使用十分重要。
一般情况下FASTA 格式并不是默认的输出格式,需要在设置中添加。
首先,打开Clustalx, 再选择Alignment -> Output Format Options ,在弹出的对话框中将FASTA format 打上勾即可。
另外,Jmodeltest 也可以使用NEXUS 格式。
下面是一个例子,假设有两条序列(虽然是个多序列比对工具,还是举个两条序列的简单例子比较容易理解):>P1ATGGGGTTTAGA TAA>P2ATGTTTAGTTAA比对之后存储的FASTA 结果应该是:>P1ATGGGGTTTAGA TAA>P2- - - ATGTTTAGTTAA注意事项:A. 输出时记得要对输出的文件名进行修改,否则会把原来的文件替换掉;B. 进行比对时,比对文件必须放在纯英文的路径下,否则软件无法读取;2. JModeltest 的使用:JModeltest 下载下来后不需要再安装,直接运行即可。
使用起来也简单易懂。
首先,点击File -> Load DNA alignment ,读取比对结果的FASTA 格式文件文件,之后选择需要进行测试的模型,点击Analysis -> Compute likelihood scores ,弹出对话框:对话框提供了4 种不同模式进行计算,每种模式包含的模型具体如下:3 schemes: JC, HKY and GTR.5 schemes: JC, HKY, TN, TPM1, and GTR.7 schemes: JC, HKY, TN, TPM1, TIM1, TVM and GTR.11 schemes: JC, HKY, TN, TPM1, TPM2, TPM3, TIM1, TIM2, TIM3, TVM and GTR.选择好这后就可以点击开始计算。


多序列比对是对三个或更多个生物学序列进行比对的过程,用于识别序列之间的相似性、保守性区域和进化关系。
以下是一些常见的多序列比对方法:
1. **CLUSTAL系列:**
- **CLUSTALW:** 是最常用的多序列比对工具之一,利用序列的相似性来构建多序列比对。
- **CLUSTAL Omega:** 是CLUSTALW的后续版本,具有更快的计算速度和更好的准确性。
2. **MAFFT:** 是一种快速而准确的多序列比对方法,利用快速傅里叶变换算法和迭代方法来处理大规模序列。
3. **T-Coffee(Tree-based Consistency Objective Function for alignment Evaluation):** 结合序列比对和序列质量评估的算法,可以整合多种信息源进行比对。
4. **MUSCLE:** 是一种高效的多序列比对工具,适用于大规模序列的比对,通常速度较快。
5. **ProbCons:** 使用概率建模进行序列比对,尤其擅长于对高度不同的序列进行比对。
6. **PASTA(Progressive Alignment of Sub-optimized Multiple Sequence Alignments):** 通过不同子集的序列构建多次比对,然后将它们集成成一个更全面的比对。
7. **Kalign:** 是一种快速的多序列比对工具,利用互信息矩阵来找到相似的序列片段。
这些方法在算法、效率和适用范围上各有特点,选择适合你研究的方法取决于序列数据的规模、相似性和所需的比对准确性。
常常,为了获得更准确的结果,研究者会结合多种方法或者使用不同参数运行同一方法以进行比较和验证。

C l u s t a l x多重序列比对图解教程(B y R a i n d y) 本帖首发于Raindy'blog软件简介:CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
ClustalX为进行多重序列和轮廓比对和分析结果提供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征。
窗口上面的下拉菜单可让你选择传统多重比对和轮廓比对需要的所有选项。
主要功能:你可以剪切、粘贴序列以更改比对的顺序;你可以选择序列子集进行比对;你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx1.81版链接地址:ist&ID=7435(请完整复制)应用:Clustalx比对结果是构建系统发育树的前提实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方的菜单栏选择“File”-“LoadSequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是否替换现有序列(Replaceexistingsequences),根据具体情形选择操作。
图1图22.编辑序列:对标尺(Ruler)上方的序列进行编辑操作,主要有Cutsequences(剪切序列)、Pastesequences(粘贴)、SelectAllsequences(选定所有序列),ClearsequenceSelection(清除序列选定)、Searchforstring(搜索字串)、RemoveAllgaps(移除序列空位)、RemoveGap-OnlyColumns(仅移除选定序列的空位)图33.参数设置:可以根据分析要求设置相对的比对参数。

名词解释:Consensus sequence:共有序列,指多种原核基因启动序列特定区域内,通常在转录起始点上游-10及-35区域存在一些相似序列。
1、FASTA序列格式:是将DNA或者蛋白质序列表示为一个带有一些标记的核苷酸或者氨基酸字符串,大于号(>)表示一个新文件的开始,其他无特殊要求。
2、Similarity相似性:是直接的连续的数量关系,是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比列的高低。
3、genbank序列格式:是GenBank 数据库的基本信息单位,是最为广泛的生物信息学序列格式之一。
该文件格式按域划分为4个部分:第一部分包含整个记录的信息(描述符);第二部分包含注释;第三部分是引文区,提供了这个记录的科学依据;第四部分是核苷酸序列本身,以“//”结尾。
4、模体(motif):短的保守的多肽段,含有相同模体的蛋白质不一定是同源的,一般10-20个残基。
5、查询序列(query sequence):也称被检索序列,用来在数据库中检索并进行相似性比较的序列。
6、打分矩阵(scoring matrix):在相似性检索中对序列两两比对的质量评估方法。
包括基于理论(如考虑核酸和氨基酸之间的类似性)和实际进化距离(如PAM)两类方法。
7、空位(gap):在序列比对时,由于序列长度不同,需要插入一个或几个位点以取得最佳比对结果,这样在其中一序列上产生中断现象,这些中断的位点称为空位。
8、PDB:PDB中收录了大量通过实验(X射线晶体衍射,核磁共振NMR)测定的生物大分子的三维结构,记录有原子坐标、配基的化学结构和晶体结构的描述等。
PDB数据库的访问号由一个数字和三个字母组成(如,4HHB),同时支持关键词搜索,还可以FASTA程序进行搜索。
9、Prosite:是蛋白质家族和结构域数据库,包含具有生物学意义的位点、模式、可帮助识别蛋白质家族的统计特征。

Clustalx 多重序列比对图解教程(By Raindy)本帖首发于Raindy'blog,转载请保留作者信息,谢谢!欢迎有写生物学软件专长的战友,加入生信教程写作群:,接头暗号:你所擅长的生物学软件名称软件简介:CLUSTALX-是CLUSTAL多重序列比对程序的Windows版本。
Clustal X为进行多重序列和轮廓比对和分析结果提供一个整体的环境。
序列将显示屏幕的窗口中。
采用多色彩的模式可以在比对中加亮保守区的特征。
窗口上面的下拉菜单可让你选择传统多重比对和轮廓比对需要的所有选项。
主要功能:你可以剪切、粘贴序列以更改比对的顺序;你可以选择序列子集进行比对;你可以选择比对的子排列(Sub-range)进行重新比对并可插入到原始比对中;可执行比对质量分析,低分值片段或异常残基将以高亮显示。
当前版本:1.83PS:如果你是新手或喜欢中文界面,推荐使用本人汉化的Clustalx 1.81版链接地址::ist&ID=7435(请完整复制)应用:Clustalx比对结果是构建系统发育树的前提实例:植物呼肠孤病毒属外层衣壳蛋白P8(AA序列)为例流程:载入序列―>编辑序列―>设置参数―>完全比对―>比对结果1.载入序列:运行ClustalX,主界面窗口如下所图(图1),依次在程序上方的菜单栏选择“File”-“Load Sequence”载入待比对的序列,如图2所示,如果当前已载入序列,此时会提示是否替换现有序列(Replace existing sequences),根据具体情形选择操作。
图1图22.编辑序列:对标尺(Ruler)上方的序列进行编辑操作,主要有Cut sequences(剪切序列)、Paste sequences(粘贴)、Select All sequences(选定所有序列),Clear sequence Selection(清除序列选定)、Search for string(搜索字串)、Remove All gaps(移除序列空位)、Remove Gap-Only Columns(仅移除选定序列的空位)图33.参数设置:可以根据分析要求设置相对的比对参数。
Using ClustalX for multiple sequence alignmentJarno TuimalaDecember 2004All rights reserved. The PDF version of this leaflet or parts of it can be used in Finnish universities as course material, provided that this copyright notice is included. However, this publication may not be sold or included as part of other publications without permission of the publisher.IndexIndex (3)Quick Start (4)1. Open ClustalX (4)2. Read in the FastA-formatted sequences (4)3. Modify the output format option, if necessary (5)4. Create an alignment (5)Creating the input file for multiple sequence alignment (6)Multiple alignment theory (7)Getting the data into ClustalX (8)Setting up the alignment parameters (9)Pairwise alignment parameters (9)Multiple alignment parameters (11)Alignment output-format (12)Creating the alignment (13)Writing alignment as Postscript (14)Assessing the quality of the alignment (15)Advanced alignment strategies (16)Advanced options (17)Do alignment from the guide tree (17)Profile alignment (17)Using secondary structure information in the profile alignment (19)Quick StartIn order to make a multiple sequence alignment using ClustalX, you should have your sequences in FastA format. If you do not know haw to do this, check the chapter “Creating the input file for multiple sequence alignment”.1. Open ClustalXAfter starting ClustalX, and you will see a window that looks something like the one below.2. Read in the FastA-formatted sequencesPull down the File-menu, and choose Load Sequences menu item. Navigate to the folder (subdirectory) that contains the input file (text-file containing the sequences in FastA-format), and choose that file. Sequences should appear in the ClustalX window.The left pane (in the figure above) lists the sequences according to the name that follows “>” symbol in the input file. The right pane shows the beginning of each sequence. You can scroll to the right to see the rest of each sequence by using the scroll bar at the bottom of the pane.3. Modify the output format option, if necessaryBefore aligning the sequences, you should make sure the output format options (from menu Alignment -> output format options) are set correctly. If you’d like to continue with phylogenetic analysis using Phylip package, you should select PHYLIP format. Note, that you should always save the Clustal formatted sequence alignment, also. Here’s an example of the output format option settings:4. Create an alignmentIn order to make the actual alignment, select “Do complete alignment” from the menu Alignment. At that point ClustalX asks for output file names. Your sequence alignment is automatically saved in those files once the alignment is ready. After the alignment has been successfully calculated, a new view will appear, and it might look something like that:Now that the alignment has been created, you can close ClustalX, and use the generated alignment files in other programs.Creating the input file for multiple sequence alignmentHere, ClustalX is going to be used for sequence alignment. It, like any other computer program requires the data it manipulates (the input file) to be in a format it can recognize. You can use your favourite word processor to create the input file, but I use Notepad.In the previous chapters, we pasted the found sequences into the text editor. Often the unedited files look like this:gi|15146064|gb|AY040893.1| Homo sapiens individual VP37 mitochondrial control region GGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGTCTGGGGGGTGTGCA CGCGATAGCATTGCGAGACGCTGGAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTC ATCCTGTTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACCTACTAAAGTGTGTTAATTAATT AATGCTTGTAGGACATAATAATAACAATTG gi|15146065|gb|AY040894.1| Homo sapiens individual VP5 mitochondrial control region GGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGTCTGGGGGGTATGCA CGCGATAGCATTGCGAGACGCTGGAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTC ATCCTATTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACTTACTAAAGTGTGTTAATTAATTClustalX can recognize several formats for the sequences, but we will use the FastA format, because that’s the one most easily downloaded from the databanks. The FastA format can be recognized, because the first line begins with the “>” character. This line contains the title of the sequence. The sequence will start from the next line. The “>” character should be followed by one word (only letters or numbers and _), which ClustalX will use as the name for the sequence in the multiple alignment that it creates. Clustal treats everything between “>” and the first space as the sequence name. I suggest you to save the original title, and just enter the new name (up to 10 characters but not more) for the sequence and one space after that. This way you can still save the Genbank accession number in the same file as the sequences. You will need the accession number, if you’re ever going to publish your results, so save them!For the sake of clarity, we will put a blank line after the first sequence before the second sequence. The order of the sequences in the file is not important.After these formatting procedures, the aforementioned sequences should look like this.>hs_vp37 gi|15146064|gb|AY040893.1| Homo sapiens individual VP37 mitochondrial control GGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGTCTGGGGGGTGTGCA CGCGATAGCATTGCGAGACGCTGGAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTC ATCCTGTTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACCTACTAAAGTGTGTTAATTAATT AATGCTTGTAGGACATAATAATAACAATTG>hs_vp5 gi|15146065|gb|AY040894.1| Homo sapiens individual VP5 mitochondrial control GGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGTCTGGGGGGTATGCA CGCGATAGCATTGCGAGACGCTGGAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTC ATCCTATTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACTTACTAAAGTGTGTTAATTAATTSave the sequences in this FastA-format as a plain text file (also known as ASCII). In the current Word version XP, the sequences should be saved as plain text, and then the encoding should be changed to MS-DOS in order to make sure that the file works in every system.Multiple alignment theoryDynamic programming can be used to align multiple sequences also. It creates an optimal alignment, but cannot be used for more than five or so sequences because of the calculation time. Therefore, progressive method of multiple sequence alignment is often applied.Clustal performs a global-multiple sequence alignment by the progressive method. The steps include:a) Perform pair-wise alignment of all the sequences by dynamic programmingb) Use the alignment scores to produce a phylogenetic tree by neighbor-joiningc) Align the multiple sequences sequentially, guided by the phylogenetic treeThus, the most closely related sequences are aligned first, and then additional sequences and groups of sequences are added, guided by the initial alignments to produce a multiple sequence alignment showing in each column the sequence variations among the sequences.Sequence contributions to the multiple sequence alignment are weighted according to their relationships on the predicted evolutionary tree. Weights are based on the distance of each sequence from the root. The alignment scores between two positions of the multiple sequence alignment are then calculated using the resulting weights as multiplication factors.As more sequences are added to the profile, gaps accumulate and influence the alignment of further sequences. Clustal calculates gaps in a novel way designed to place them between conserved domains. Gaps found in the initial alignments remain fixed. New gaps are then introduced into the multiple alignment when more sequences are added, but gaps can never be deleted, only added. Clustal also implements methods, which try to compensate for the scoring matrix (e.g., PAM), expected number of gaps, and differencies in sequence length.Clustal has advanced options:a) Add sequences with weightb) Add weights to different sequence positionsc) Add a sequence or alignment to an alignmentd) Use user-defined tree for alignmentSome of these will be discussed in the next chapters.The problem with progressive alignment is the dependence of the ultimate multiple sequence alignment on the initial pair-wise alignments. The very first sequences to be aligned are the most closely related on the sequence tree. If these sequences align very well, there will be few errors in the initial alignments. However, the more distantly related these sequences, the more errors will be made, and these errors will be propagated to the multiple sequence alignment. A second problem with the progressive alignmentmethod is the choise of suitable scoring matrices and gap penalties that apply to the set of sequences.Getting the data into ClustalXStart ClustalX, and you will see a window that looks something like the one below.Pull down the File-menu, and choose Load Sequences menu item. Navigate to the folder (subdirectory) that contains the input file (text-file containing the sequences in FastA-format), and choose that file.The left pane (in the figure above) lists the sequences according to the name that follows “>” symbol in the input file. The right pane shows the beginning of each sequence. You can scroll to the right to see the rest of each sequence by using the scroll bar at the bottom of the pane.Setting up the alignment parametersThe alignment is done is several succeeding steps: (from Clustal documentation)1. Reset All Gaps (Alignment->Alignment parameters, Edit->Remove all Gaps)2. Refine Pairwise Alignment Parameters (Alignment->Alignment parameters)3. Refine Multiple Alignment Parameters (Alignment->Alignment parameters)4. Refine Output Format Options (Alignment->Output Format Options)5. Write Alignment as Postscript (File->Write Alignment as Postscript)6. Assess the quality of the alignmenta. Not satisfied -> Go to step 1.b. Satisfied -> Refine the alignment by handPairwise alignment parametersIn order to create the pairwise alignment, ClustalX needs to know what penalties to assign for the creation of a gap and for the extension of that gap. Choose Pairwise Alignment Parameters from the Alignment-menu. You will see a dialog box like the one below.The first choise, Pairwise Alignments, allows you to choose between Slow-Accurate and Fast-Approximate methods. The Slow-method is preferred, but if you are aligning so many sequences or the sequences are so long that the program takes a long time to run,you may want to use the Fast-method. The Fast-method uses a k-tuple method for pairwise alignment, whereas Slow-method uses a full dynamic programming algorith. The box shows the default values for Gap Opening and Gap Extension. Decreasing the gap penalties will allow the introduction of more gaps, and less mismatches. This may result is matches that do not reflect homology (identity by descent). Increasing gap penalties wil have an opposite effect, but may result in missing matches that actually are homologies.Weight matrix parameters can be changed too. The IUB DNA weight matrix scores matches as 1.9 and mismatches as 0, except that it scores all X’s and N’s as matches to any IUB ambiguity symbols. The Protein Weight Matrices are equivalent to the same matrices used as evolutionary models in the production of the dendrogram. All the matrices have their strenghts and down-sides: PAM has been used for years, but is now somewhat outdated, and the Gonnet can be more appropriate for your purposes. BLOSUM seems to be best for searching databases.You can create and load in your own matrix into ClustalX. For the description of the file format, take a look at the file matrices.h in the ClustalX-folder:For amino acids:char *amino_acid_order = "ABCDEFGHIKLMNPQRSTVWXYZ";short blosum30mt[]={4,0, 5,-3, -2, 17,0, 5, -3, 9,0, 0, 1, 1, 6,-2, -3, -3, -5, -4, 10,0, 0, -4, -1, -2, -3, 8,-2, -2, -5, -2, 0, -3, -3, 14,0, -2, -2, -4, -3, 0, -1, -2, 6,0, 0, -3, 0, 2, -1, -1, -2, -2, 4,-1, -1, 0, -1, -1, 2, -2, -1, 2, -2, 4,1, -2, -2, -3, -1, -2, -2, 2, 1, 2, 2, 6,0, 4, -1, 1, -1, -1, 0, -1, 0, 0, -2, 0, 8,-1, -2, -3, -1, 1, -4, -1, 1, -3, 1, -3, -4, -3, 11,1, -1, -2, -1, 2, -3, -2, 0, -2, 0, -2, -1, -1, 0, 8,-1, -2, -2, -1, -1, -1, -2, -1, -3, 1, -2, 0, -2, -1, 3, 8,1, 0, -2, 0, 0, -1, 0, -1, -1, 0, -2, -2, 0, -1, -1, -1, 4,1, 0, -2, -1, -2, -2, -2, -2, 0, -1, 0, 0, 1, 0, 0, -3, 2, 5,1, -2, -2, -2, -3, 1, -3, -3, 4, -2, 1, 0, -2, -4, -3, -1, -1, 1, 5,-5, -5, -2, -4, -1, 1, 1, -5, -3, -2, -2, -3, -7, -3, -1, 0, -3, -5, -3, 20,0, -1, -2, -1, -1, -1, -1, -1, 0, 0, 0, 0, 0, -1, 0, -1, 0, 0, 0, -2, -1,-4, -3, -6, -1, -2, 3, -3, 0, -1, -1, 3, -1, -4, -2, -1, 0, -2, -1, 1, 5, -1, 9, 0, 0, 0, 0, 5, -4, -2, 0, -3, 1, -1, -1, -1, 0, 4, 0, -1, -1, -3, -1, 0,2,4}; /*For the DNA:char *nucleic_acid_order = "ABCDGHKMNRSTUVWXY";short clustalvdnamt[]={10,0, 0,0, 0, 10,0, 0, 0, 0,0, 0, 0, 0, 10,0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 10,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 10,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0};Multiple alignment parametersChoose Multiple Alignment Parameters from the Alignment-menu. A new dialog box will appear.The pairwise and multiple alignment options are set independently, because Clustal needs to know both. As have been already discussed, a matrix of pairwise alignments is first calculated, and on the basis of those distances, a multiple alignment is formed. Using different settings for these alignment steps gives us more flexibility to affect how the alignment is done.Compared to pairwise alignment there are a couple of new settings. Delay Divergent Sequences determines how different two sequences must be in order for their alignment to be delayed. This tries to compensate for the bias introduced by progressive alignment method.DNA transition weight can be modified. Weight 0 means that transitions are scored as mismatches, and weight 1 mean that transitions are given the same weight as transversions. For distantly related DNA sequences, the weight should be near to zero; for closely related sequences it can be useful to assign a higher score.You don’t have to choose the individual Weight Matrix any more, because we know how dissimilar the sequences are by now. ClustalX will automatically choose the most appropriate matrix inside one matrix series. So, if you changed the matrix series to be used in the pairwise alignment options, be sure to change it here too.Alignment output-formatThe last thing to be modified before performing the alignment is the output-format. When ClustalX creates an alignment it writes the aligned sequences into a file. There are different multiple alignment formats, which might be needed depending on what program you have planned to use for further analyses.For the phylogenetic construction using Phylip-package, choose the Phylip as an output format. You should also always remember to write the aligned sequences in the Clustal-format, because it is convenient for both publishing the alignment and refining the alignment in the Notepad. If you plan to publish the sequences, turn the sequence numbering on.You can modify the output-format by selecting the Output Format Options menu item form the Aligment-menu.Creating the alignmentFinally, it is time to create the alignment. Choose the Do Complete Alignment under the Alignment menu.ClustalX tells you what it is doing, but it usually locks the computer, so do not plan to use the computer for any other purposes while the alignment is still under construction.One word of warning, though. ClustalX does not understand spaces in the names of the folders. Therefore, for example, My Documents, cannot be used in the path.After the alignment is done, the main window will be updated with the aligned sequences.The bases are colored, which makes the assessment of alignment that much easier. The histogram (or line) below the ruler indicates the degree of similarity. Peaks indicate positions of high similarity, and valleys positions of low similarity.The grey line just above the sequences is used to mark strongly conserved positions. The “*” character indicates positions that have been fully conserved.Writing alignment as PostscriptIt is possible to use the files we have just created to construct a phylogenetic tree, but the quality and the value of that tree will be no better than the quality of the alignment, and we have not yet considered quality.It is also important to understand that no matter how dissimilar the sequences are, ClustalX will always produce an alignment. The mere existence of the alignment does not mean that the sequences are related. It is up to the user to ensure that the sequences in the data set are actually homologous and therefore alignable.The quality of the alignment is easier to check if there exists hard copies of the sequences. You have two possibilities. You might print the ClustalX-format alignment, which exists in the text-format, and use that, but then you’ll lose the information of the colors.The other possibility is to install Ghostscript and Ghostview programs, which allow the user to manipulate Postscript documents, and print the alignment as postscript, which includes the colors.Go to File-emu and select Write Alignment as Postscript. A dialog box opens.You have some options to change, but after you are satisfied with them, click OK. Open the postscript-file in Ghostview. The Ghostview-program might give you some warnings about the incorrect type of the postscript-file, but ignore them by clicking on the Cancel-button.Now you can print this alignment by selecting the printer-button in the left and top corner of the window.Assessing the quality of the alignmentIn practice, many alignments are produced, and they are compared together. You can start by using the default ClustalX options for pairwise and multiple alignment options. In the next alignments try lowering and highering the gap options by 50%. Produce, say 10-20 different alignments, and then compare those together. Often you need to try more than 10 different settings in order to find the best alignment. What you also need to keep in mind is that alignment is not an absolute thing. It is a best guess according to some algorithm used by a computer program or by an experienced human eye. It is necessary for the user to carefully examine each alignment to see if it makes biological sense.After printing the alignments you need to examine them to see if most of the gaps make sense. If many of the gaps seem to be arbitrary (i.e., you think you could have done better by eye), then you need to improve the alignment. If there are large regions that are present in only one or two sequences, you may need to delete those regions from the alignment. In practice, most of the programs used for phylogenetic tree reconstruction, will delete all the column containing any gaps from the analysis, so you don’t need to bother.There are some guidelines on how to assess the quality. First remember, that we want to maximize similarity, and minimize dissimilarity. Therefore, the number of gaps is one parameter you should pay attention to. Use the histogram in the bottom of the alignment and the “*” characters above it to assess the conservation of different areas of the alignment.It is also biologically unsound to assume that there are many gaps with equal spaces between them. Usually gaps are clustered, and are more common in certain areas than in other areas. So, look for the number and length of the conserved blocks of columns. If the pattern of the gaps looks like they have been randomly inserted, choose another alignment. This is, of course, assuming that the sequences are relatively closely related. If you are aligning an area where there are no functional genes, the above’s all you can do. If you have some knowledge on the functional regions of the sequence you are aligning, you can use this information when assessing the quality of the alignment.The functional regions are often more or less conserved between the relatively closely related sequences. Therefore, quite a few gaps should be inserted into those areas, and most of the gaps should be inserted into less well conserved areas, for example, in the spacer regions between alfa-helixes.Although it is very time consuming, attempting to improve the alignment through this process of examination and modification of penalties is probably the single most important thing you can do to ensure a high-quality alignment and make a high-quality phylogeny estimation possible.Advanced alignment strategiesIf you have very difficult sequences to align, you can try iterative alignment procedure in order to get a better estimate of the real alignment. First, produce an initial alignment with some quite closely fitting parameter values. Then, produce a new alignment from this initial alignment without removing gaps before this second alignment. You can iterate the alignment using the same settings, but doing the alignment based on the previous alignment multiple times. Sooner or later the alignment will stabilize, and will not change anymore with the same parameters. This is the best, “iterated”, alignment for those settings.Normally, it is important to reset the gaps before producing the alignment with new settings, but this is not done with iterative alignment.You can also realign only a part of the sequences. Hold down the left mouse button, and paint a selection. Then, from the Alignment menu select Realign Selected Residue Range. ClustalX will then do the alignment using the current setting only for the selected residues. You can also produce a new alignment for selected sequences (Realign Selected Sequences). Sequences can be selected by holding down the left mouse-button, and then dragging downwards in the sequence name list.Advanced optionsDo alignment from the guide treeIf you have some data on the relationships of the sequences, you can construct a tree in the Newick-format, and use that for producing a multiple sequence alignment. Produce a tree like the one below:((Pan_verus1:0.02428,Pan_verus2:0.01474):0.03203,(Pan_velle1:0.00437,Pan_velle2:0.00579):0.01402,(Pan_trog1:0.00306,Pan_trog2:0.00306):0.05015);Save the tree in a file in text-format.In Clustal, select the Do Alignment From Guide Tree from the Alignment menu. This way you can try to get around the difficulties of the progressive alignment, which might create wrong alignments, if the sequences are very divergent. This can also be used to combine morphological data into the sequence alignment step: the knowledge of the relationships of the taxons can be used to guide the alignment process.Profile alignmentProfile alignment is used for a couple of purposes, but we first discuss how to align a new sequence into an existing alignment.There is a pull-down menu on the main window top left-hand corner. From the menu change to Profile Alignment Mode. The Alignment view window is now split into two parts. The upper part contains the alignment we just created, and lower is empty. The upper part is called “profile 1” and the lower part is “profile 2”.In the File-menu you have the options to load profiles 1 and 2. Now we already have the profile one available, so we only need to load the profile 2. Let’s do that. The main window is updated, and look something like the one below.From the alignment-menu, select first the option “Align Sequences to Profile 1”. After that select “Align Profile 2 to Profile 1”. This will create an alignment file, where all the sequences are together. Note that you should always first align the sequences in the profile 1 before aligning those (already aligned) sequences to the profile 2!This is a very handy way to add new sequences into an existing alignment. Otherwise you would have needed to calculate the initial alignment again, which could have been very laborous in the case of many or very long sequences.Using secondary structure information in the profile alignmentAs has been shortly discussed above the sequence alignment can be guided by the secondary structure of the protein, if such information is available. Often such alignments are more biologically plausible than the alignments done “randomly”.In ClustalX the gap penalties are raised at core alpha helix (A) or beta strand (B) residues. The structure information can be used only in the Profile Alignment Mode. These gap penalties cannot be used in the multiple alignment mode. There are two ways to include structure information in Clustal, but here we present only the easier one, which describes the domain areas of the protein. Then the penalties are adjusted in the ClustalX dialog box.First we need to create the input files. In the first input file, which the first sequence of all the sequences to aligned, a descriptions of the domains (helix or strand) is included. The second input file contains the rest of the sequences in the FastA-format.The information about the domains is most easily acquired from the SWISS-PROT descriptions. Find the relevant information from /swissprot/, and after you have acquired the SRS results, click on the Accession Number link on the top of the page. This will take you to the plain text description.From the description find the lines starting with two capital letters: ID, FT, SQ, and the sequence. Copy those lines into a text file (i.e., into Notepad) and save the file. It should now something like the one below.ID XRC1_HUMAN STANDARD; PRT; 633 AA.FT HELIX 315 403 BRCT 1.FT HELIX 538 629 BRCT 2.SQ SEQUENCE 633 AA; 69525 MW; 30CC2421345ABFC2 CRC64;MPEIRLRHVV SCSSQDSTHC AENLLKADTY RKWRAAKAGE KTISVVLQLE KEEQIHSVDI GNDGSAFVEV LVGSSAGGAG EQDYEVLLVT SSFMSPSESR SGSNPNRVRM FGPDKLVRAA AEKRWDRVKI VCSQPYSKDS PFGLSFVRFH SPPDKDEAEA PSQKVTVTKL GQFRVKEEDE SANSLRPGAL FFSRINKTSP VTASDPAGPS YAAATLQASS AASSASPVSR AIGSTSKPQE SPKGKRKLDL NQEEKKTPSK PPAQLSPSVP KRPKLPAPTR TPATAPVPAR AQGAVTGKPR GEGTEPRRPR AGPEELGKIL QGVVVVLSGF QNPFRSELRD KALELGAKYR PDWTRDSTHL ICAFANTPKY SQVLGLGGRI VRKEWVLDCH RMRRRLPSRR YLMAGPGSSS EEDEASHSGG SGDEAPKLPQ KQPQTKTKPT QAAGPSSPQK PPTPEETKAA SPVLQEDIDI EGVQSEGQDN GAEDSGDTED ELRRVAEQKE HRLPPGQEEN GEDPYAGSTD ENTDSEEHQE PPDLPVPELP DFFQGKHFFL YGEFPGDERR KLIRYVTAFN GELEDYMSDR VQFVITAQEW DPSFEEALMD NPSLAFVRPR WIYSCNEKQK LLPHQLYGVV PQAThe ID line gives description of the sequence and what it codes. The FT lines describe what kind of domains are present in the protein. Those should say either HELIX or STRAND (always double-check those; they might be inaccurate). The SQ line gives molecular weight and some other information of the succeeding amino acid sequence.For checking the secondary structures, go to http://www.embl-heidelberg.de/predictprotein/submit_def.html and paste in the first protein sequence. In a short while the results will be emailed to you. From the results, you’ll find a description: AA |MPEIRLRHVVSCSSQDSTHCAENLLKADTYRKWRAAKAGEKTISVVLQLEKEEQIHSVDI|PHD sec | EEEEEEEEEE HHHHHHHHH HHHHHHHHHHH EEEEEE EEEEE |Rel sec |993678997772578764999998651136878999946883699885235453354451|detail:prH sec |000000000000000116899988764467888899962100000000001111000000|prE sec |006778898875210000000000000000000000000003799886431212566664|prL sec |993210001114688872000001225432111000027886200012557665322225|subset: SUB sec |LL.EEEEEEEE.LLLLL.HHHHHHHH...HHHHHHHH.LLL.EEEEEE..L.L..E..E.|accessibility3st: P_3 acc | eebebebbbbbbeeebeebbeebbebee ebebbeeeeebbbbbbebeeeeebbbbeb|10st: PHD acc |397060600000099707600770070774570600777770000006067776000060|Rel acc |002325036846201120058314433100121164412342639850325320404504|subset: SUB acc |.....b..bbbb.......bb..bb.........bbe...e.b.bbb...e...b.bb.b|The line marked with AA gives the original sequence, and the the next line, starting with PHD sec gives the predicted secondary structures. Rel sec gives the reliability of the secondary strusture prediction. H mean helix, and E mean sheet. You can use this information for the description of the structures for ClustalX input.You have just created the first input file. The second file should include the other sequences you’re interested in in FastA-format (see above).After preparing these input files, go to ClustalX, and switch to Profile Alignment Mode. From the File menu select Load Profile 1, and search for the first input file. If ClustalX recognizes the file to contain the weights for the gaps it asks you whether to use the penalties or not.If you have formatted the files as described above, but ClustalX does not recognize the weights, go to Alignment->Alignment Parameters->Secondary Structure parameters. From the opening dialog box turn the Use Profile Secondary Structure option on (yes). After that try to load the first input file again.。
序列比对之Clustalx与Clustalw使用指南这几天实验需要做多序列比对,很久不做了,一时之间不知道如何使用clustal这个工具了。
在网上搜集了一些资料,做个整理,总结了Clustalx和Clustalw的使用,省得以后久不使用又生疏了,又要去整理了,在此分享给大家,希望有所帮助。
1.先提供下载地址:官方下载地址:/download/current/Clustalx、Clustalw的各种最新版本都能下载到,包括linux、Win、Mac...2.原理:序列同源性分析:是将待研究序列加入到一组与之同源,但来自不同物种的序列中进行多序列同时比较,以确定该序列与其它序列间的同源性大小。
这是理论分析方法中最关键的一步。
完成这一工作必须使用多序列比较算法。
常用的程序包有CLUSTAL等;Clustal是一个单机版的基于渐进比对的多序列比对工具,由Higgins D.G.等开发。
有应用于多种操作系统平台的版本,包括linux版,DOS版的clustlw,clustalx等。
CLUSTAL是一种渐进的比对方法,先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树,对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止。
3.操作3.1 Clustalx的操作第一步:输入序列文件。
第二步:设定比对的一些参数。
参数设定窗口。
第三步:开始序列比对。
第四步:比对完成,选择保存结果文件的格式--------------------------------------------3.2 Clustalw的使用(一)第一步:按屏幕提示选择1,输入序列文件注意:请把你需要比对的多条序列合并为一条,放在一个文件中第二步:选择保存文件的形式,按提示选择,你会的!第三步:参数设置好后,按Enter返回主界面,开始比对!EBI提供的在线clustalw服务/clustalw/可以在这里得到更多关于clustal的帮助:http://www-igbmc.u-strasbg.fr/BioInfo/ClustalX/Top.html。
生物信息学工具介绍1、FASTA[10](/fasta33/)和BLAST[11](http://www.nc /BLAST/)是目前运用较为广泛的相似性搜索工具。
比较和确定某一数据库中的序列与某一给定序列的相似性是生物信息学中最频繁使用和最有价值的操作。
本质上这与两条序列的比较没有什么两样,只是要重复成千上万次。
但是要严格地进行一次比较必定需要一定的耗时,所以必需考虑在一个合理的时间内完成搜索比较操作。
FASTA使用的是Wilbur-Lipman 算法的改进算法,进行整体联配,重点查找那些可能达到匹配显著的联配。
虽然FASTA不会错过那些匹配极好的序列,但有时会漏过一些匹配程度不高但达显著水平的序列。
使用FASTA和BLAST,进行数据库搜索,找到与查询序列有一定相似性的序列。
一般认为,如果蛋白的序列一致性为25-30%,则可认为序列同源。
BLAST(Basic Loc al Alignment Search Tool,基本局部联配搜索工具)是基于匹配短序列片段,用一种强有力的统计模型来确定未知序列与数据库序列的最佳局部联配。
BLAST 是现在应用最广泛的序列相似性搜索工具,相比FASTA 有更多改进,速度更快,并建立在严格的统计学基础之上。
这两个工具都采用局部比对的方法,选择计分矩阵对序列计分,通过分值的大小和统计学显著性分析确定有意义的局部比对。
BLAST根据搜索序列和数据库的不同类型分为5种:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
实验六:多序列比对- Clustal、MUSCLE西北农林科技大学生物信息学中心实验目的:学会使用Clustal 和MUSCLE 进行多序列比对分析。
实验内容:多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似区域只能通过将多个序列同时比对才能识别。
只有在多序列比对之后,才能发现与结构域或功能相关的保守序列片段,而两两序列比对是无法满足这样的要求的。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守motif 的搜寻等具有非常重要的作用。
我们这节课主要学习两个广泛使用的多序列比对软件-Clustal、MUSCLE。
一、Clustal/Clustal 是一种利用渐近法(progressive alignment)进行多条序列比对的软件。
即先将多个序列两两比较构建距离矩阵,反应序列之间的两两关系;随后根据距离矩阵利用邻接法构建引导树(guide tree);然后从多条序列中最相似(距离最近)的两条序列开始比对,按照各个序列在引导树上的位置,由近及远的逐步引入其它序列重新构建比对,直到所有序列都被加入形成最终的比对结果为止(Figure 6.1)。
Clustal 软件有多个版本。
其中Clustalw 采用命令行的形式在DOS 下运行;Clustalx 是可视化界面的程序,方便在windows 环境下运行;Clustal omega 是最新的版本,优点是比对速度很快,可以在短短数小时内比对成百上千的序列,同时由于采用了新的HMM 比对引擎,它的比对准确性也有了极大的提高,有DOS 命令行和网页服务器版。
我们今天主要学习clustalx 的使用。
范例1. 采用clustalx 进行多序列比对。
生物信息学实验教程实验一、基因、蛋白质序列分析【实验目的】1、掌握基因、蛋白质序列检索的操作方法;2、熟悉蛋白质基本性质分析及其电子表达谱3、蛋白基因的引物设计【实验内容】1、使用Entrez或SRS信息查询系统检索人脂联素(adiponectin)蛋白质序列;2、使用网站对上述蛋白质序列进行分子质量、氨基酸组成、和疏水性等基本性质分析;3、蛋白基因的引物设计【实验方法】1、人脂联素基因、蛋白质序列的检索:(1)调用Internet浏览器并在其地址栏输入Entrez网址(/Entrez);(2)在Search后的选择栏中选择nucleartide\protein;(3)在输入栏输入homo sapiens adiponectin;(4)点击go后显示序列接受号及序列名称;(5)点击序列接受号NP_004788 (adiponectin precursor; adipose most abundant genetranscript 1 [Homo sapiens])后显示序列详细信息;(6)将序列转为FASTA格式保存(参考上述步骤使用SRS信息查询系统检索人脂联素蛋白质序列);(7)进入UNIGENE数据库分析其电子表达谱2、进入网站对人脂联素蛋白质序列进行分子质量、氨基酸组成和疏水性等基本性质分析:3、利用prime prime5.0设计此基因PCR引物4、独立完成NYGGF4、LYRM1两个基因的上述操作。
【作业】1、提交使用上述软件对人脂联素、NYGGF4、LYRM1蛋白质序列进行基本性质分析及其电子表达谱蛋白质实验二、序列结构预测【实验目的】1、熟悉基于序列同源性分析的蛋白质功能预测,了解基于motif、结构位点、结构功能域数据库的蛋白质功能预测;2、了解蛋白质结构预测。
【实验内容】1、对人脂联素蛋白质序列进行基于NCBI/Blast软件的蛋白质同源性分析;2、对人脂联素蛋白质序列进行motif结构分析;3、对人脂联素蛋白质序列进行二级结构和三维结构预测。