美国及欧洲药典系统适应性要求
- 格式:pdf
- 大小:546.00 KB
- 文档页数:22

盐酸缬更昔洛韦C14H22N6O5·HCl 390.82L-缬氨酸,9-[[2-羟基-1-(羟甲基)乙氧基]甲基]鸟嘌呤酯,盐酸盐L-缬氨酸,2-[(2-氨基-1,6-二氢-6-氧代-9H-嘌呤-9-yl)甲氧基]-3-羟丙基酯,盐酸盐[175868-59-5]按干燥品计算,本品含C14H22N6O5·HCl应为97.0%~102.0%。
包装与贮存——密封包装,25℃保存,允许温度浮动范围15℃~30℃。
鉴别A:红外吸收(197K)B:紫外吸收(197U)溶液:10μg/ml溶剂:甲醇C:本品的水溶液(1→20)显氯化物的鉴别反应。
(191)水分,方法一(921):取本品100mg,依法检查,含水分不得过8.0%。
炽灼残渣(281):取本品1g,依法检查,遗留残渣不得过0.10%。
重金属,方法一(231):不得过0.002%。
异丙醇内标溶液——取100μL的1,4-二氧六环到100mL容量瓶,用二甲基甲酰胺定容。
标准储备液——分别取1.0mL异丙醇与0.1mL甲苯到同一100mL容量瓶,用二甲基甲酰胺定容。
(注:甲苯用来调整系统适用性)标准溶液——取2.0mL内标溶液置于反应瓶中,再加入100μL标准储备液,混匀,待用。
系统适用性溶液——使用标准溶液供试品溶液——精密称取90~100mg的盐酸缬更昔洛韦,置于反应瓶中,精密量取2.0mL的内标溶液加入其中,混匀待用。
色谱系统(见色谱法(621))——配有氢火焰离子化检测器的气相色谱仪,色谱柱为涂布3.0-µm G43固定相的0.53mm× 30m毛细管柱,载气为氦气,流速为10.5mL/min,分流比为1:15。
色谱程序设定如下:初始柱温为40℃,保持10min,后在25℃/min下逐渐升温至240℃。
(注:每次进样后调节柱温至240℃,并保持15min左右)进样口温度保持在250℃,检测器温度保持在300℃。

<1225>V ALIDATION OF COMPENDIAL PROCEDURES药典规程的验证Test procedures for assessment of the quality levels of pharmaceutical articles are subject to various requirements. According to Section 501 of the Federal Food, Drug, and Cosmetic Act, assays and specifications in monographs of the United States Pharmacopeia and the National Formulary constitute legal standards. The Current Good Manufacturing Practice regulations [21 CFR 211.194(a)] require that test methods, which are used for assessing compliance of pharmaceutical articles with established specifications, must meet proper standards of accuracy and reliability. Also, according to these regulations [21 CFR 211.194(a)(2)], users of analytical methods described in USP-NF are not required to validate the accuracy and reliability of these methods, but merely verify their suitability under actual conditions of use. Recognizing the legal status of USP and NF standards, it is essential, therefore, that proposal for adoption of new or revised compendial analytical procedures be supported by sufficient laboratory data to document their validity.用于评价药物质量水平的测试规程受到多种要求的影响。

浅析EP(欧洲药典)、USP(美国药典)和CP(中国药典)的质量标准区别名词解读:EP: European Pharmacopeia欧洲药典,缩写为EP。
为欧洲药品质量检测的指导文献。
所有药品或原料的生产厂家在欧洲范围内销售和使用的过程中,必须遵循《欧洲药典》的质量标准。
USP:U.S. Pharmacopeia 美国药典,缩写为USP。
为美国药品质量检查的指导文献。
出版时为U.S. Pharmacopeia / National Formulary《美国药典/国家处方集》(简称USP/NF)。
CP:The People's Republic of China Pharmacopoeia中华人民共和国药典,缩写CP。
为中国制药行业中药、西药、生物制品等质量检查的指导文献,分一部:中药、二部:西药、三部:生物制品。
2010版将于10月1日起执行。
正文:由于其质量标准中药品的检测项目繁多,我就不一一来比较,只拿出相对较为复杂的“液相法含量测定”来进行简单的比较。
知识水平有限、时间也不是很多,因此他们之间的许多不同可能没有发掘出来,还请大家和我一道,继续发掘、一起分析!EP和USP在一定程度是相通的,也就是说他们对于同一个药品的质量标准是相同的,但也不能排除有不同的情况,比如在流动相比例、检测波长上就有的品种不同。
而他们对于CP来说,区别就很多了,我要描述的也主要是这个方面:色谱柱:CP一般的规定是填料,而对柱长、粒径等不做规定。
那么,对我们而言,色谱柱的选择就成了一个问题,我们需要花时间去找柱子。
而EP、USP,基本上就把柱子的长度、直径、粒径都规定好了。
洗脱方法:EP、USP多采用梯度洗脱,配制起来比较复杂,稍微的ph值不适当或比例问题就会影响实验结果,且在标准中大多规定了主峰或相关杂质峰的保留时间,因此难度较大。
而CP多采用等度洗脱,操作、试验都相对较容易。
无论是梯度还是等度,只要把相关的物质分离出来就可以了。



美国usp 800标准美国usp 800标准是指美国药典委员会(USP)发布的一项关于药品在医疗保健环境中的安全使用和管理的标准。
该标准主要针对医疗保健机构内的药品管理和药品安全,特别是针对有毒药物的管理。
这些有毒药物可能对医护人员和患者造成危害,因此需要严格的管理和控制。
根据美国usp 800标准,医疗保健机构需要建立一套完善的有毒药物管理系统,包括采购、接收、存储、配制、分发、使用和废弃等环节。
这些环节需要严格的操作规程和管理措施,以确保有毒药物不会对人员和环境造成危害。
在采购环节,医疗保健机构需要选择可靠的供应商,确保采购的药品符合质量标准,并且具有相关的许可证和资质。
在接收环节,需要对药品进行验收,并及时处理异常情况。
在存储环节,需要建立专门的存储区域,对有毒药物进行单独存放,并严格控制温度、湿度和光线等环境因素。
在配制、分发和使用环节,需要严格按照操作规程进行操作,并使用个人防护装备,以降低职业暴露的风险。
在废弃环节,需要对废弃药品进行正确处理,以防止对环境造成污染。
美国usp 800标准的实施,对医疗保健机构提出了更高的要求,需要加强对有毒药物管理的重视,加强对医护人员的培训和教育,加强对药品管理的监督和检查。
只有通过严格的管理和控制,才能有效地保护医护人员和患者的安全,减少药品管理中的风险和事故发生。
总的来说,美国usp 800标准的实施,对于提高医疗保健机构的药品管理水平,保障医护人员和患者的安全,具有重要的意义。
医疗保健机构应当认真学习和贯彻这一标准,加强内部管理,提高工作人员的安全意识,建立健全的管理制度,确保有毒药物的安全使用和管理。
同时,相关部门应当加强监督和检查,及时发现和纠正存在的问题,推动美国usp 800标准的全面实施,为医疗保健环境的安全和健康作出贡献。

美国药典USP-FAQ-CompliancewiththeUSP-NF英中150828Compliance with the USP-NF (药典符合性)1. What does compliance with USP–NF standards mean?An article of commerce that is recognized in the USP–NF complies with USP–NF standards when it meets all of the requirements stated in the article’s monograph, applicable General Chapters, and the General Notices (with monograph requirements superseding those of the General Chapters and General Notices, in any cases where requirements differ). Applicable standards apply at all times in the life of an article, from production to expiration. Thus, any official article is expected to meet the compendial standards if tested, and any official article actually tested as directed in the relevant monograph must meet such standards to demonstrate compliance. Frequency of testing and sampling are left to the preferences or direction of those performing compliance testing, and other users of USP–NF, including manufacturers, buyers, or regulatory authorities. (General Notices, section3.10) 问:什么是USP-NF标准的符合性?答:一个市售药品如果完全符合USP-NF中相应各论、适用通则,凡例的要求(若各论的要求与通则和凡例的要求不一致时,则以各论的要求为准),则该药品符合USP-NF标准。
系统适应性——美国药典系统适应性是气相和液相色谱分析方法的重要组成部分,用于证明色谱系统的分离度和重现性能满足样品的分析要求。
测试基于这样的原理:仪器、电路、方法和样品组成一个整体系统,我们可以对这个系统进行测试评估。
影响色谱系统的因素包括:●流动相的组成、离子强度、温度和pH值●柱子大小、流速、柱温和压力●固定相特点,包括填料类型,载体形状、粒径、孔径、表面积等。
●常用固定相为反相硅胶,以十八碳烷基健合硅胶最常用,其它经过化学修饰的硅胶也有使用。
分离度R s是理论塔板数n的函数(也叫柱效),α是分离因子,k是容量因子(所有符号的意义见前文“色谱定义和说明”部分)。
在规定的色谱条件下,n表示洗脱物中相邻化合物的分离程度,可作为衡量色谱系统柱效能的指标,但是不如直接测试的结果可靠。
峰的尖锐程度部分反映柱效,这个参数对检查微量物质至关重要。
标准品或者标准溶液需要重复进样以确保精密度。
除非个论中有规定, 系统适用性五针的数据的相对标准偏差不超过2.0%, 如果超过2.0%的话, 需要进样六针。
在含量测定中,如果纯品含量100%,则相对标准偏差没有最大值限制,这个值可根据多次进样对照溶液来计算:%RSD=KB/t90%,n-1K为常数0.349,由公式k=(0.6/)×(t90%,5/)计算得来,表示B=1.0时六次进样的相对标准偏差。
B是个案中规定的上限。
n是对照溶液的进样次数(3≤n≤6),t90%,n-1是自由度为n-1、置信水平为90%,双侧检验时的t 值。
除非另有规定,RSD不能超过下表中的值。
此规定不适用于相关物质检测。
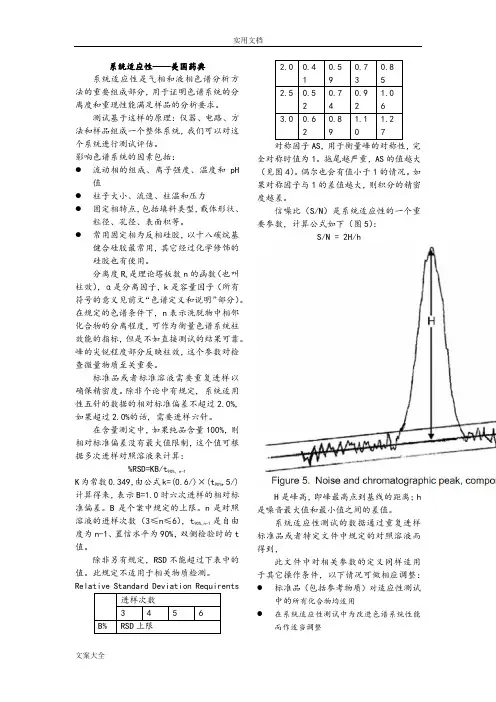
Relative Standard Deviation Requirents对称因子AS,用于衡量峰的对称性,完全对称时值为1。
拖尾越严重,AS的值越大(见图4)。
偶尔也会有值小于1的情况。
如果对称因子与1的差值越大,则积分的精密度越差。
信噪比(S/N)是系统适应性的一个重要参数,计算公式如下(图5):S/N = 2H/hH是峰高,即峰最高点到基线的距离;h 是噪音最大值和最小值之间的差值。

•2398•前持诗件耐元Drug Evaluation Research第43卷第12期2020年12月【审评规范]欧盟和美国对药品说明书【适应症】项目的撰写要求及启示萧惠来国家药品监督管理局药品审评中心,北京100022摘要:介绍欧盟和美国有关药品说明书【适应症】项目撰写的法规和指导原则,特别是最近欧洲药品管理局(EMA)发布的“治疗适应症的用语”。
从这些管理文件得到的启示是相关法规规定不宜太粗糙,应具体、精准,应包括内容和格式的规定;而且要有指导原则伴随,以保证法规落到实处。
熟悉掌握药品说明书【适应症】项目的撰写要求对规范撰写其他项目有普遍指导意义,对于药品说明书的监管也有裨益。
关键词:药品说明书;适应症项目;法规;指导原则中图分类号:R951文献标志码:A文章编号:1674-6376(2020)12-2398-06DOI:10.7501/j.issn.l674-6376.2020.12.008Requirements of EU and United States for writing indication section of labeling and its enlightenmentXIAO HuilaiCenter for Drug Evaluation,National Medical Products Administration,Beijing100022,ChinaAbstract:This paper introduces the regulations and guidelines on the writing of indication section of drug labeling in the EU and the United States,especially the Wording of therapeutic indication published by EMA recently.The Enlightenment from these management documents is that the relevant regulations should not be too rough,they should be specific and accurate,and should include the provisions of content and format,and should be accompanied by guidelines to ensure the implementation of regulations. To be familiar with the writing requirements of indication section of drug labeling is of general guiding significance for standardizing the writing of other sections,and also beneficial to the supervision of drug labeling.Key words:drug labeling;indication section;regulation;guideline药品说明书是传递合理用药信息的重要工具,也是医师、药师、护师和患者治疗用药时的科学依据,还是药品生产、供应部门向医药卫生人员和普通民众宣传介绍药品特性、指导合理、安全用药和普及医药知识的主要媒介。
浅析EP(欧洲药典)、USP(美国药典)和CP(中国药典)的质量标准区别名词解读:EP: European Pharmacopeia欧洲药典,缩写为EP。
为欧洲药品质量检测的指导文献。
所有药品或原料的生产厂家在欧洲范围内销售和使用的过程中,必须遵循《欧洲药典》的质量标准。
USP:U.S. Pharmacopeia 美国药典,缩写为USP。
为美国药品质量检查的指导文献。
出版时为U.S. Pharmacopeia / National Formulary《美国药典/国家处方集》(简称USP/NF)。
CP:The People's Republic of China Pharmacopoeia中华人民共和国药典,缩写CP。
为中国制药行业中药、西药、生物制品等质量检查的指导文献,分一部:中药、二部:西药、三部:生物制品。
2010版将于10月1日起执行。
正文:由于其质量标准中药品的检测项目繁多,我就不一一来比较,只拿出相对较为复杂的“液相法含量测定”来进行简单的比较。
知识水平有限、时间也不是很多,因此他们之间的许多不同可能没有发掘出来,还请大家和我一道,继续发掘、一起分析!EP和USP在一定程度是相通的,也就是说他们对于同一个药品的质量标准是相同的,但也不能排除有不同的情况,比如在流动相比例、检测波长上就有的品种不同。
而他们对于CP来说,区别就很多了,我要描述的也主要是这个方面:色谱柱:CP一般的规定是填料,而对柱长、粒径等不做规定。
那么,对我们而言,色谱柱的选择就成了一个问题,我们需要花时间去找柱子。
而EP、USP,基本上就把柱子的长度、直径、粒径都规定好了。
洗脱方法:EP、USP多采用梯度洗脱,配制起来比较复杂,稍微的ph值不适当或比例问题就会影响实验结果,且在标准中大多规定了主峰或相关杂质峰的保留时间,因此难度较大。
而CP多采用等度洗脱,操作、试验都相对较容易。
无论是梯度还是等度,只要把相关的物质分离出来就可以了。
2020年欧盟及美国对口罩等防疫用品的准入要求按照中央应对新冠疫情工作领导小组部署和国务院联防联控机制会议要求,为深化国际疫情防控合作,加强医疗物资出口质量的监管,商务部会同海关总署、药监局于2020年3月31日发布了《关于有序开展医疗物资出口的公告》,要求出口的检测试剂、医用口罩、医用防护服、呼吸机、红外体温计等五类产品,必须取得我国医疗器械产品注册证书,符合进口国(地区)的质量标准要求,海关凭药监部门批准的医疗器械产品注册证书验放。
1.医疗物资质量认证不能盲目在中国,质量认证大致分成两大类,一类是强制性认证,一类是自愿性认证。
医用口罩、防护服、呼吸机,在我国都属于药品监督管理部门规定必须符合医疗器械注册管理的产品,并不属于强制性认证管理范围。
相关企业一定要找合法的认证机构。
中国现在经过批准的有600多家认证机构,从事产品、服务、管理体系等认证,这些信息在国家认监委的网站上都可以查询到。
企业如果要做认证,一定要找具有相应合法资质的机构。
2.医疗物资进欧盟市场得加贴CE欧盟CE“认证”,不叫CE认证制度,实际上是一种CE标志的准入制度,按照欧盟规定,列入CE标志管理制度中的产品,必须加贴CE标志之后,才能进入到欧盟市场进行销售。
证明这个产品符合了CE标志要求的评价手段主要有两方面:对绝大部分在CE标志管理制度内的产品,企业采取符合性自我声明的方式,按照相关程序要求,你自己能证明产品符合相关要求,就可以加贴CE标志,进入到欧盟市场;但是也有一些风险性比较高的产品,必须获得欧盟授权的公告机构认证,经过他们的认证,这个产品才能加贴CE标志。
另外,现在网上也有谈美国FDA的“认证”,实际上FDA是美国食品药品监督管理局,它开展的所谓“认证”并不是一种认证评价活动,而是一种行政许可性质的制度,实质上是政府的注册管理。
3.欧盟CE证书可以花钱买到?市场监管总局对认证活动依法进行监管,对出现的违法案件进行严厉打击。
欧盟EUGMP标准要求中文版欧盟药品管理规则第 4 卷药品生产质量管理规范1998 版前言欧洲共同体制药工业在药品的开发,生产和控制过程中保持高标准的质量保证。
上市许可系统保证由有能力的权威机构对药品的安全,质量和有效性是否达到相应的规定进行评估。
生产许可系统保证在欧洲市场上获准销售的药品是由授权的生产商生产,其日常活动由权威机构定期检查。
无论是在欧共体之内销售,还是在欧共体之外销售,所有欧共体的药品生产企业都必须通过生产许可。
有两个药品生产和质量管理指导原则,药品生产和质量管理规范(GMP)和指南来源于两个指导原则, 一个是人用药物指导原则(指导原则 91/356/EEC)一个是兽用药物指导原则(指导原则91/412/EEC),这两个指导原则1991年被欧共体采纳。
根据这些原则,制定了详细的药品生产和质量管理规范,用于对申请生产许可的企业进行评估和对药品生产企业进行检查的基础。
GMP的原则和详细的指南适用于需要按照第16条75/319/ EEC 和修改的第24条81/851/EEC要求认证的所有的操作。
也与所有其它大规模药品生产过程,诸如医院负责的临床试验用药的制备有关。
所有的成员国和工业企业本身都同意GMP适用于人用药物的生产,也适用于兽用药物的生产。
在两个附录中对兽用药品和兽用免疫药品的GMP指南做了详细的调整。
指南用章来表述,每章用标题来概括章节的原则内容。
第一章质量管理列出了药品生产的质量保证的基本概念。
后续各章的原则列出了质量保证的目标和提供了足够的让生产商在执行这一原则时所必须考虑的基本要素。
这一指南除了在9个章节中表述了GMP的基本要素外, 还包括一系列附录提供了与之有关的活动的特定范围的细节。
有时几个附录同时使用,如关于无菌制剂,辐射性药物,生化药物的附录。
在附录后还列出了这一指南所使用的术语表.指南的第一版在 1989 年出版, 包括一个无菌药品生产的附录。
第二版在1992 年1月出版; 欧共体指到原则包括给人用药品和兽用药品的GMP提供原则和指南的欧共体于1991 年6月 13 日颁布的91/356指导原则和1991 年7月23 日颁布的91/412指导原则。
系统适应性——美国药典系统适应性是气相和液相色谱分析方法的重要组成部分,用于证明色谱系统的分离度和重现性能满足样品的分析要求。
测试基于这样的原理:仪器、电路、方法和样品组成一个整体系统,我们可以对这个系统进行测试评估。
影响色谱系统的因素包括:●流动相的组成、离子强度、温度和pH值●柱子大小、流速、柱温和压力●固定相特点,包括填料类型,载体形状、粒径、孔径、表面积等。
●常用固定相为反相硅胶,以十八碳烷基健合硅胶最常用,其它经过化学修饰的硅胶也有使用。
分离度R s是理论塔板数n的函数(也叫柱效),α是分离因子,k是容量因子(所有符号的意义见前文“色谱定义和说明”部分)。
在规定的色谱条件下,n表示洗脱物中相邻化合物的分离程度,可作为衡量色谱系统柱效能的指标,但是不如直接测试的结果可靠。
峰的尖锐程度部分反映柱效,这个参数对检查微量物质至关重要。
标准品或者标准溶液需要重复进样以确保精密度。
除非个论中有规定, 系统适用性五针的数据的相对标准偏差不超过2.0%, 如果超过2.0%的话, 需要进样六针。
在含量测定中,如果纯品含量100%,则相对标准偏差没有最大值限制,这个值可根据多次进样对照溶液来计算:%RSD=KB/t90%,n-1K为常数0.349,由公式k=(0.6/)×(t90%,5/)计算得来,表示B=1.0时六次进样的相对标准偏差。
B是个案中规定的上限。
n是对照溶液的进样次数(3≤n≤6),t90%,n-1是自由度为n-1、置信水平为90%,双侧检验时的t 值。
除非另有规定,RSD不能超过下表中的值。
此规定不适用于相关物质检测。
Relative Standard Deviation Requirents对称因子AS,用于衡量峰的对称性,完全对称时值为1。
拖尾越严重,AS的值越大(见图4)。
偶尔也会有值小于1的情况。
如果对称因子与1的差值越大,则积分的精密度越差。
信噪比(S/N)是系统适应性的一个重要参数,计算公式如下(图5):S/N = 2H/hH是峰高,即峰最高点到基线的距离;h 是噪音最大值和最小值之间的差值。
<1225>V ALIDATION OF COMPENDIAL PROCEDURES药典规程的验证Test procedures for assessment of the quality levels of pharmaceutical articles are subject to various requirements. According to Section 501 of the Federal Food, Drug, and Cosmetic Act, assays and specifications in monographs of the United States Pharmacopeia and the National Formulary constitute legal standards. The Current Good Manufacturing Practice regulations [21 CFR 211.194(a)] require that test methods, which are used for assessing compliance of pharmaceutical articles with established specifications, must meet proper standards of accuracy and reliability. Also, according to these regulations [21 CFR 211.194(a)(2)], users of analytical methods described in USP-NF are not required to validate the accuracy and reliability of these methods, but merely verify their suitability under actual conditions of use. Recognizing the legal status of USP and NF standards, it is essential, therefore, that proposal for adoption of new or revised compendial analytical procedures be supported by sufficient laboratory data to document their validity.用于评价药物质量水平的测试规程受到多种要求的影响。
中国、美国、欧洲药典对HPLC方法的规定所谓药典方法,顾名思义,药典专论所收载的方法,但是否每个人都知道,药典方法考虑到各个实验室的差异,有一定的可调范围。
下面我们具体的来探讨一下这个问题吧!一、中国药典2015规定0512高效液相色谱法规定如下:品种正文项下规定的条件除填充剂种类、流动相组分、检测器类型不得改变外,其余如色谱柱内径与长度、填充剂粒径、流动相流速、流动相组分比例、柱温、进样量、检测器灵敏度等,均可适当改变,以达到系统适用性试验的要求。
调整流动相组分比例时,当小比例组分的百分比例X 小于33%时,允许改变范围为0.7X 〜1.3X ; 当X大于33%时,允许改变范围为X —10%〜X+ 10%。
由上文可知,中国药典专论方法除填充剂种类、流动相组分(比例调整在规定范围内)、检测器类型不得改变外,其他条件改变了,还是认为是药典方法。
但中国药典没有规定方法确认之说,所以,即使是中国药典方法,也是应该按分析方法验证的相关规定进行全套的方法验证。
二、美国药典41规定USP41 <621> CHROMATOGRAPHY 规定如下:为符合系统适用性的要求而调整的操作条件是允许的,除非专论项下另有规定,下面为所列的最大的可调范围,这些调整需要额外的确认数据。
为确认新条件对方法的适用性,评估调整对相关分析性能特征存在的潜在影响。
多条件的调整,将对系统性能产生累积影响,实施前需仔细考虑。
1、pH of mobile phase(HPLC): 流动性缓冲液的pH:±0.2 units,适用梯度及等度。
2、Concentration of salts in buffer (HPLC): ±10%(pH允许情况下),适用梯度及等度。
3、Ratio of componentsin mobile phase (HPLC):流动相中组分(≤50%)可以在此比例上再调节±30%,且任一组分比例的调节不应超过±10%(相对于总的流动相),含三组分的流动相的调节可以分组分进行,含双组分的流动相的调节可在最小的组分进行,双组份和三组分的调节实例如下:双组分:例如流动相50:50,其中一组分50%的30%是15%,但是它超过了占总体比例±10%的规定,所以这个流动相比例的调节范围应以±10%为限,可以在不超过40:60~60:40的范围之间调节。