活性基团聚合
- 格式:doc
- 大小:190.00 KB
- 文档页数:6

活性聚合的优点和用途活性聚合是一种重要的聚合物材料,具有许多优点和广泛的应用。
下面将详细介绍活性聚合的优点和用途。
活性聚合的主要优点如下:1. 高反应活性:活性聚合物具有高度活跃的载体和活化基团,从而可以促进聚合过程中的高效反应。
这种高反应活性可以使聚合物分子得以在较短时间内形成,提高生产效率。
2. 多样的合成方式:活性聚合易于通过不同的聚合方法进行合成,如自由基聚合、阴离子聚合、阳离子聚合、开环聚合等。
这种多样性使得活性聚合物可以制备出多种不同结构和性质的聚合物材料。
3. 可控的分子结构:活性聚合可以通过控制反应条件和使用合适的催化剂来实现对聚合物的分子结构的精确控制。
例如,可以调控聚合物分子量、分子量分布、支化程度等。
这种可控性使得活性聚合物能够满足各种特定应用的需求。
4. 优异的性能:由于活性聚合过程的高反应活性和可控性,所得到的聚合物材料通常具有良好的性能表现。
例如,活性聚合物可以具有高度交联的结构,从而具有优异的耐热性、耐溶剂性和物理力学性能。
5. 容易修饰功能化:活性聚合物可以通过在聚合过程中引入特定的功能单体来实现聚合物的功能化修饰。
通过这种方式,可以为聚合物材料赋予特定的性能,如抗菌性、降解性、生物相容性等。
活性聚合的应用非常广泛,以下列举了几个常见的应用领域:1. 高分子材料:活性聚合物广泛应用于高分子材料的制备。
通过控制活性聚合过程,可以获得具有特定物理和化学性质的聚合物材料,如聚乙烯、聚苯乙烯、聚丙烯等。
这些高分子材料在塑料、橡胶、纺织、包装等领域得到广泛应用。
2. 功能性材料:活性聚合可以用于制备具有特殊功能的材料。
例如,通过引入含有活性基团的单体,可以在聚合过程中实现对聚合物链的控制修饰,从而得到具有特定功能的材料。
如荧光标记材料、生物相容性材料、抗菌材料等。
3. 涂料和胶黏剂:活性聚合物被广泛用于制备涂料和胶黏剂。
由于活性聚合的可控性和多样性,可以制备出不同性质和功能的涂料和胶黏剂。



ATRP活性聚合1背景ATRP是最近几年发展起来的,由Matyjaszewski课题组发现。
王锦山和Matyjaszewski川采用1一氯代苯乙烷作为引发剂,氯化亚铜和联吡啶形成的络合物作为催化剂,在130℃下引发苯乙烯本体聚合,反应3h产率可达95%,聚合物的分子质量分布在1.5以下,理论分子质量和实验值符合较好。
原子转移自由基聚合(ATRP)兼具自由基聚合与活性聚合的特点,适用单体范围广ATRP作为一种新颖的精确聚合反应,能实现可控/活性聚合,产物可达到预期的分子量,且分子量分布较窄,因此是大分子设计的有效工具。
许多烯类单体已成功地用ATRP合成出结构确定的均聚物、无规共聚物、交替共聚物、梯形共聚物、嵌段/接枝共聚物和新型聚合物刷、梳形聚合物、星形聚合物、树枝状聚合物及有机/无机杂化材料。
2机理及组成典型的原子转移自由基聚合的基本原理如图1.1所示:在引发阶段,处于低氧化态的转移金属卤化物(盐)M t n从有机卤化物R—X中吸取卤原子X,生成引发自由基R.发单体聚合,形成链自由基R—M n·,R—M n·可从高氧化态的金属络合物M t n+1-X中重新夺取卤原子而发生钝化反应,形成R-M n-X,并将高氧化态的金属卤化物还原为低氧化态M t n,即:始终伴随着一个自由基活性种与有机大分子卤化物休眠种的可逆转移平衡反应正是由于M t n/ M t n+1催化剂的氧化和还原过程,能使体系保持一个很低的自由基浓度,大大减少了自由基之间的终止反应。
1引发剂ATRP引发剂一般是碳上具有诱导或者共轭结构的R—X,卤素基团必须能够快速、选择性地在增长链和转移金属之间交换,快引发对控制聚合物低分散系数很重要此外R.x引发剂引发ATRP之后,经过增长反应可以得到端基很明确的聚合物.也就是引发剂可以被引入到聚合物的两端.这样,通过设计合成带有小同基团的引发剂,经过ATRP反应后实现对高分子链末端功能化或生成高分子反应中间体,如大分子单体、遥爪聚合物、大分引发剂等.另外引发剂的片段还可以作为标记基团引入聚合物,对聚合物进行跟踪分析,进一步功能化得到特殊性能高分子及分子组装等2单体理论上讲大部分的可以进行自由基聚合单体都可以进行ATRP聚合,但是必须要求能找到合适的引发剂,催化剂,配体。

基团转移聚合反应及其应用基团转移聚合反应,简称“ATRP”,是一种通过控制聚合反应的速率和分子量,制备高分子材料的方法。
ATRP方法在有机合成和高分子材料制备方面具有广泛的应用,对制备功能材料具有重要的作用。
ATRP反应的基本原理是通过引入活性基团,利用化学键断裂的自由基反应,来引发分子间的链扩增反应。
在ATRP反应中,引入的基团通常是卤素、硝基、苯基等活性基团。
这些基团与金属催化剂形成稳定的配合物,完成活化反应。
ATRP反应的优点在于可以控制聚合物的分子量和分子量分布。
通过控制反应条件可以调整反应速率和分子量,使得高分子材料的结构和性能可以被精确定制。
ATRP反应的另一个优点是可以被用来制备复杂的高分子结构,如星形高分子、网状高分子等。
ATRP反应在材料科学、有机合成等领域得到了广泛应用。
ATRP反应产生的高分子具有特殊的性质,这使得它们在药物传递、化妆品、电子材料、纳米结构的制备等多个领域具有潜在的应用价值。
在材料科学领域,ATRP反应已经成功地应用于制备嵌段共聚物、电子界面材料、光学材料、传感器和纳米材料。
总之,ATRP反应是一种高度可控的聚合反应,具有许多优点,可以被用于制备复杂的高分子结构,产生具有特殊性质和应用价值的高分子材料。
未来,这种反应将继续在材料科学和有机合成领域得到广泛应用。


活性聚合活性聚合(Living Polymerization)摘要:活性聚合(Living Polymerization)是一种特殊的聚合反应方法,可以在反应过程中控制聚合物的分子量和分子量分布。
活性聚合反应中的聚合物链可以在不与其他链发生反应的情况下不断延长,使得聚合物具有更高的结构控制性和功能化潜力。
本文将介绍活性聚合的基本原理、常见的活性聚合方法以及其在材料科学和工业中的应用。
1. 活性聚合的基本原理活性聚合是一种通过控制聚合物的生长速率和反应活性来实现的聚合过程。
与传统的自由基聚合不同,活性聚合是一种具有可逆性和控制性的反应,其中单体分子通过与活性种子发生反应而聚合,而活性种子可以通过适当的反应条件进行控制。
这种可控的聚合方式使得聚合物的结构和性质具备更高的可调性和定制性。
2. 常见的活性聚合方法2.1 原子转移自由基聚合(ATRP)原子转移自由基聚合是一种常见的活性聚合方法,可以以较高的控制度合成具有规则结构和可控分子量的高分子。
在ATRP中,通过引入适当的转移剂(如卤代烷烃)和催化剂(如铜络合物),可以实现聚合物链的生长和停止。
这种方法适用于各种单体,如甲基丙烯酸酯、丙烯酸酯和丙烯酸等,可以用于合成聚合物的共聚物和嵌段共聚物。
2.2 硅醚聚合(SIP)硅醚聚合是一种在低温条件下进行的活性聚合方法,它通过引入硅醚链传递剂来控制聚合物的生长和反应速率。
硅醚链传递剂可以在聚合反应中引发传递反应,从而实现聚合链的延长和停止。
这种方法可用于合成线性和星形共聚物,如聚乳酸-聚乙二醇嵌段共聚物。
2.3 离子液体-金属有机框架催化剂聚合(IL-MOFs)离子液体-金属有机框架催化剂聚合是一种新兴的活性聚合方法,可以通过引入具有催化活性的离子液体-金属有机框架催化剂来控制聚合反应。
这种方法在聚合物链的生长和停止过程中具有高度的可控性和选择性,并且可以用于合成精确结构和多功能聚合物。
3. 活性聚合的应用3.1 材料科学领域活性聚合在材料科学领域具有广泛的应用,可以合成具有精确结构和控制形态的聚合物。



活性聚合特点及应用活性聚合是一种特殊的聚合反应,其特点是在聚合过程中活化单体,使其在聚合链上生成活性中间体,然后再将其他单体加入到活性中间体上进行进一步的聚合反应。
相比传统的聚合方法,活性聚合具有以下几个特点:1. 进行聚合反应的单体通过活化可以控制聚合的速度、分子量和分子量分布。
通过控制活化单体的种类和活化程度,可以实现精确控制单体的反应活性,从而控制聚合速度。
而通过控制聚合反应时间和反应温度,可以控制聚合的分子量和分子量分布。
2. 活性聚合可以实现单体的逐步添加,从而使得聚合反应更加灵活。
传统的聚合方法中,所有单体都被同时加入到反应体系中,导致聚合链的延伸速度很快,很难控制聚合反应的速度和分子量。
而活性聚合中,单体可以逐个加入到活性中间体上,可以根据需要控制聚合的速度和分子量。
3. 活性聚合可以实现多种功能单体的共聚反应。
活性中间体具有较高的反应活性,可以与多种单体反应生成复杂的共聚物结构。
通过控制反应条件和单体的添加顺序,可以实现对共聚物结构的调控,从而获得具有特定功能的材料。
4. 活性聚合可以在室温下进行,反应条件温和。
传统的聚合反应通常需要高温或特殊催化剂的作用,而活性聚合反应可以在室温下进行,反应条件温和,有利于提高材料的质量。
活性聚合在材料科学和化学工程领域有着广泛的应用。
以下是一些常见的应用领域:1. 开发新型聚合物材料。
活性聚合方法可以实现对聚合物结构的精确控制,可以合成出具有特定性能的材料。
例如,通过活性聚合反应可以合成具有特定分子量和分子量分布、表面活性基团、孔隙结构的聚合物材料,用于制备纳米材料、吸附剂等。
2. 设计功能性聚合物。
活性聚合反应可用于合成具有特殊功能的聚合物材料,例如温度敏感性、光敏感性、磁敏感性等。
这些聚合物可以应用于药物缓释、智能传感器、生物材料等领域。
3. 制备微纳制品。
活性聚合反应可以在微纳米尺度上控制聚合物的形态和结构,从而实现定向生长、模板刻蚀等制备微纳米结构。
聚合物的聚合机理在化学领域中,聚合物是由许多重复单元通过共价键相互连接而成的大分子。
这些分子通常由单体通过聚合反应得到,而聚合反应的过程正是聚合物形成的关键。
聚合物的聚合机理是指在聚合过程中单体如何发生化学反应,如何相互连接以及如何形成高分子链的过程。
聚合反应的第一步是单体的引发,通常通过引发剂或催化剂的作用,单体中的双键或活性基团被激活,使其具有活性,从而能够发生反应。
在引发之后,单体之间发生加成反应或缩合反应,形成聚合物的主链。
加成反应是指单体分子中的双键开启,与其他单体分子中的双键或活性基团结合,形成共价键,使聚合物主链逐渐延伸;而缩合反应则是指两个单体分子中的活性基团结合,释放小分子,形成新的共价键。
在聚合过程中,引发剂的作用至关重要。
引发剂可以分为自由基引发剂、离子引发剂和配位引发剂三类。
自由基引发剂通过引发自由基聚合反应,是最常见的引发剂类型。
在自由基引发剂作用下,单体中的双键或活性基团断裂,形成自由基,自由基之间进行反应并连续地将单体分子连接在一起,逐渐形成聚合物链。
离子引发剂则通过引发阴离子或阳离子聚合反应,而配位引发剂则通过配位活化单体中的分子,促使其发生聚合反应。
聚合反应的速率和选择性取决于引发剂的种类、单体的结构以及反应条件等因素。
在实际应用中,需要选择适合的引发剂和反应条件,以实现所需聚合物的合成。
此外,聚合物的分子结构和性能也受聚合反应的影响,合成过程中需要控制聚合物链的长度、分子量分布以及支化程度,以满足特定应用的要求。
总的来说,聚合物的聚合机理是一个复杂而精密的过程,涉及引发剂、单体结构、反应条件等多方面因素。
通过深入理解聚合反应的机理,可以精确控制聚合过程,合成出具有特定结构和性能的聚合物,为材料科学和化工领域的发展提供重要支持。
1。
活性/可控自由基聚合在20世纪50、60年代,自由基聚合达到了它的鼎盛时期。
但由于存在链转移和链终止反应,传统自由基聚合不能较好地控制分子量及大分子结构[1]。
1956年美国科学家Szwarc等提出了活性聚合的概念[2],活性聚合具有无终止、无转移、引发速率远远大于链增长速率等特点,与传统自由基聚合相比能更好地实现对分子结构的控制,是实现分子设计、合成具有特定结构和性能聚合物的重要手段。
但离子型活性聚合反应条件比较苛刻、适用单体较少,且只能在非水介质中进行,导致工业化成本居高不下,较难广泛实现工业化。
鉴于活性聚合和自由基聚合各自的优缺点,高分子合成化学家们联想到将二者结合,即可控活性自由基聚合(CRP)或活性可控自由基聚合。
CRP可以合成具有新型拓扑结构的聚合物、不同成分的聚合物以及在高分子或各种化合物的不同部分链接官能团,适用单体较多,产物的应用较广,工业化成本较低。
目前实现“活性”/可控自由基聚合可分以下几种途径: (1) 稳定“活性”自由基聚合(SFRP);(2) 原子转移自由基聚合(ATRP);(3)可逆加成-断裂链转移聚合(RAFT)。
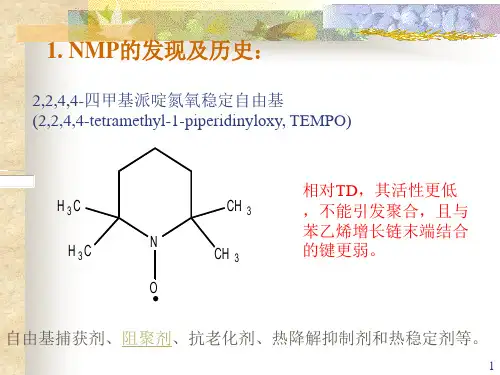
一、稳定“活性”自由基聚合(SFRP)SFRP属于非催化性体系,是利用稳定自由基来控制自由基聚合。
其机理是按照下面的可逆反应进行:外加的稳定自由基X·可与活性自由基P·迅速进行失活反应,生成“休眠种”P-X,P-X能可逆分解,又形成X·及活性种自由基P·而链增长。
有研究表明,使用烷氧胺作引发剂效果好[3]。
反应体系中的自由基活性种P·可抑制在较低的浓度,这样就可以减少自由基活性种之间的不可逆终止作用,从而聚合反应得到控制。
稳定自由基X·,主要有TEMPO(2,2,6,6-四甲基-1-哌啶氮氧自由基)和CoⅡ·,TEMPO属于稳定的有机自由基;CoⅡ·属于稳定的有机金属自由基。
四种聚合反应在化学领域,聚合反应是指将许多小分子单体通过共价键连接起来形成高分子化合物的过程。
这种反应在合成新材料、制备聚合物等方面起着至关重要的作用。
在本文中,我们将介绍四种常见的聚合反应,它们分别是加成聚合、缩聚聚合、环氧开环聚合和自由基聚合。
加成聚合加成聚合是通过单体之间发生加成反应,逐步线性生长成高分子的过程。
其中,最典型的加成聚合反应是乙烯通过开环聚合形成聚乙烯。
这种聚合反应通过引发剂或催化剂的作用,将乙烯单体分子的双键依次开启并连接起来,形成高分子链。
加成聚合反应通常需要高度纯净的单体,以避免副反应的发生。
缩聚聚合缩聚聚合是指通过两种或多种具有活性基团的单体,通过活性基团间的反应形成高分子化合物。
常见的缩聚单体有二元酸和二元胺,它们在反应时会释放小分子,如水等。
例如,聚酰胺的制备即是通过二元酸与二元胺之间的缩聚反应形成的。
缩聚聚合通常发生在具有活性基团的单体之间,反应条件相对严苛。
环氧开环聚合环氧开环聚合是指环氧化合物通过环氧环被打开,并与另外一种活性基团发生反应形成高分子的过程。
环氧开环聚合反应广泛应用于涂料、粘合剂等行业。
环氧化合物在受到引发剂或催化剂作用后,环氧环容易被开启,而后与其他单体发生反应从而形成高分子链。
环氧开环聚合反应中,控制反应条件能够得到特定结构的高分子化合物。
自由基聚合自由基聚合是通过自由基参与的聚合反应,自由基是一种带有未成对电子的中间体,对芳香烃、乙烯等具有高活性。
通过引发剂的作用,单体分子中的双键被打开并产生自由基,自由基之间会发生链转移、重组等反应,形成高分子化合物。
自由基聚合是一种重要的聚合方式,广泛应用于橡胶、塑料等材料的合成过程中。
综上所述,加成聚合、缩聚聚合、环氧开环聚合和自由基聚合是四种常见的聚合反应,它们在不同领域的应用和原理机制各有特点。
通过深入了解这四种聚合反应,可以更好地掌握聚合物合成的原理和方法,推动相关领域的发展和创新。
基团转移聚合摘要:介绍了基团转移聚合的机理和近几年来基团转移聚合在聚合单体、引发剂、催化剂、聚合实施方法等方面的新进展。
基团转移聚合技术在各种特定结构高分子合成中起到重要作用。
关键词:基团转移聚合,活性,单体,引发剂,催化剂自1956年提出活性聚合的概念以来,活性聚合技术得到迅速发展。
活性聚合技术的发展为合成结构和组成可控的聚合物材料提供了可能,使聚合物材料的应用范围进一步扩大,成为21世纪材料科学发展的基础。
目前,活性聚合方法主要有活性阴离子聚合、阳离子聚合、自由基聚合、配位聚合和基团转移聚合等[1-2]。
1983年美国杜邦公司的O.w.webster在美国化学会186次会上宣读了“基团转移聚合反应”(Group Transfer Polymerization简称GTP)“,立即在世界上引起强烈反应[3]。
GTP它是以α、β—不饱和酯、酮、酰胺和腈为单体,以带有硅、锗、锡烷基基团的化合物为引发剂,用阴离子型或路易斯酸型化合物作催化剂,选用适当的有机物作溶剂,通过催化剂与引发剂端基的硅、锗、锡原子配位,激发硅、锗、锡原子,使之与单体的羰基氧或氮结合成共价键单体中的双键完成加成反应,硅、锗、锡烷基基团移至末端形成“活性”化合物。
以上过程反复进行,得到相应的聚合物。
此项反应技术被认为是继本世纪50年代Ziegler 发现用配位催化剂使烯烃定向聚合之后的又一重要的新聚合技术。
GTP在这短短的几年里发展迅速,在控制聚合物分子量、分子量分布、端基官能化和反应条件等方面比通常的聚合方法具有更多的优越性,从而为“高分子的分子设计”又增添了新内容;同时已引用此种技术生产汽车面漆、合成液晶聚合物和一些特殊的聚合物,如嵌段、遥爪型高分子材料等。
可以预料,在不久的将来,GTP技术会取得更大的进展[4-5]。
一、GTP反应机理目前基团转移聚合中研究得最多的单体是甲基丙烯酸甲酯(MMA)和丙烯酸乙酯(EA)。
由于MMA的活性最大,因此研究得更为深入。
下面就以MMA为单体,乙烯酮的硅烷缩醛类化合物为引发剂来介绍反应机理。
1.链引发亲核性阴离子催化剂Nu-先与引发剂或活性聚合物中活性端基上的硅原子配位,使硅原子活化,然后活化的硅原子与单体中羧基氧原子相连形成六配位硅的中间过渡态,最后形成中间体的端基上连着活性基团。
引发剂与单体进行如下Michael加成反应,在端基上重新生成一个三甲基硅氧基和一个双键[6]。
2.链增长聚合过程中的每一步,都是从链末端向单体转移一个特定基团:三甲基硅基基团,起着活性传送体的作用,基团转移聚合由此得名。
这个过程反复进行,最后得到高聚物。
3.链终止在加入终止剂之前,聚合物链均含有三甲基硅氧基,具有向其它单体继续加成的能力,是活性聚合链。
为终止其活性可加入带有活泼氢的终止剂。
例如最常用的是甲醇,则获得最终需要的产物,其结构非常接近理想的模型化合物。
所以GTP与阴离子聚合一样,除非加入甲醇等活泼氢物质,链增长一直保持活性,不会终止。
因此,对整个聚合体系的纯度要求十分严格,否则容易使聚合反应提前终止,达不到预期的实验目的。
二、GTP研究新进展1、单体[7]1.1 大位阻单体在酯基结构上作文章,引入各种大位阻基团,通过GTP得到具有特殊性能的高分子是此类单体的共同特点。
如用单体1、2(见图1)可合成具有液晶性能的高聚物[8]。
甲基丙烯酸长链酯的聚合物也能通过GTP合成,但由于较大酯基的空间位阻影响反应速率,通常都要提高聚合温度。
Nakano和Puts[9]还进行了具有光学活性的大侧链高分子的合成,单体分别是3、4。
Nakano还比较了单体3在GTP、自由基、阴离子等不同方式下聚合的结果,GTP条件下产率最高,结构也最规整,再一次体现了GTP在分子结构控制上的优势。
1.2 多官能团单体另一类单体为多官能团单体,通过GTP法得到高分子后,在主链或侧链上留下可供进一步反应的官能团,成为各种功能高分子的预聚物。
如单体5、6,进行GTP后再通过高分子反应得到侧链带有羧基和羟基的聚合物,和早年利用多官能团引发剂只在链末端带有羧基和羟基的高分子不同,由于每一结构单元均带有亲水基,在表面活性剂、金属离子络合剂等高度官能团化的聚合物的控制合成上有重要意义。
Mykytiuk等将单体8聚合后再进行水解,得到用正常GTP无法得到的聚甲基丙烯酸,分子量分布很窄MW/Mn<1.1[10]。
含对阴离子及自由基聚合敏感基团的单体9在GTP条件下聚合后,得到可溶性的线型聚合物,侧链上的双键可进行交联反应制得互穿网络聚合物[11]。
双官能团单体可以进行环状加成聚合或生成梯形聚合物。
Carothers[12]等用单体11合成了结构单元中含6元环的产物,聚合过程如下另外,单体10、12也能聚合产生相应环状产物,这些环状聚合产物分子量大约在5000~10000之间,Mw/Mn在1.2~1.3之间[13]。
Sogah等用双官能团单体13合成梯形聚合物[14]。
1.3 其它特殊单体第三类为各种特殊的GTP单体,充分显示了GTP广泛的适用性。
如硅烷羟醛缩合反应(Aldol-GTP)新单体。
这种聚合方式是1986年由Sogah和Webster[15]首先发现的。
最为典型的Aldol-GTP反应如下:这种反应单体中含有“转移”基团SiR3,而不需另加含Si引发剂,这一点与普通GTP有本质区别。
产物结构单元中含OSiR3基团,水解后得到聚乙烯醇。
反应的催化剂一般用Zn 的卤化盐,其它Lewis酸如AlCl3、SnCl4等产率较低,而Zn盐中以ZnBr2分子量控制最好。
2、引发剂近来,开发的新引发剂侧重于特殊结构高分子的合成,尤其重视高分子的官能化。
引发剂上带有特殊官能团或特殊结构,可在产物末端得到相应官能团及结构。
如19、20可分别在链尾引入羰基及羟基,而21可在链尾留下双键已是众所周知,22可在链尾引入羰基,23链尾引入(OEt)2P=O或(OH)2P=O,如果用含(OEt)2P=O基团化合物中止反应,可生成两端带官能团的遥爪型聚合物。
Liu[16]等合成了侧链带有类似引发剂结构的高聚物24,并预言它能成为GTP 有趣的引发剂。
很明显,可由此制得聚有机硅氧烷为主链,聚丙烯酸酯为侧链的接枝共聚物。
3、催化剂早期的亲核阴离子型催化剂存在制备困难或在THF中溶解性差等缺点。
近几年,在催化剂方面所作的工作主要围绕提高引发效率、降低产物分子量分散性、提高实用性及降低成本这些方面展开。
例如Bandermann等发现TBA+CN-是一种良好的、溶解性能极佳的甲基丙烯酸酯与丙烯酸酯GTP催化剂[17]。
种类繁多,实用性较强的催化剂是氧阴离子,例如:脂肪芳香类的羧酸盐、酚盐、亚磺酸盐,这些催化剂性能与它们的碱性很有关系,而且能在一个较宽广的范围内催化GTP。
4、溶剂GTP反应通常是在溶剂中进行。
目的是消除聚合时产生的热量和准确配制引发剂和催化剂。
按催化剂类型可选用两类不同溶剂。
若选择阴离子催化剂时,一般宜采用给电子溶剂。
如THF、CH3CN、CH3OCH2、CH2OCH2CH3及甲苯等。
若选用Lewis酸催化剂时,则应避免用给电子体的溶剂。
因为它易与Lewis酸发生络合配位,妨碍了催化剂与单体羰基中的氧配位,所以宜用卤代烃、芳烃作溶剂。
鉴于引发剂和活性质点对水十分敏感,因此聚合所用的溶剂必须绝对无水。
三、GTP发展前景的展望GTP技术已在众多具有良好分子量及结构控制的功能高分子的合成中发挥了重要作用。
显而易见,随着GTP的发展,研究重点已不再是这个方法本身,而是如何将这一“活性”聚合的新方法用于合成各种新型的、具有特殊结构或特殊功能的高分子。
参考文献[1] MaS H,Dickre IB,Henlre W R.Aqueouspigment dispersionsoontainingABC triblockpo lmy erdispersants.EP556649.1993[2] 《基团转移聚合反应中的有机硅引发剂》[3] 《基团转移聚合反应》[4] f12]邓云祥.高分子化学物理及应用基础.北京:高等教育出版社.1997.[5] 《《高分子合成技术的新进展》》[6] 《聚合物合成反应的新进展》[7] 《《基团转移聚合新进展》》[8] 邹友思,林国良,姚青青等.高分子学报,1996,1:116[9] (1)Nakano T,Sogah D Y, J Am Chem Soc, 1995,117(1 ): 534 ~ 535; ( 2 ) Puts R D, Sogah D Y.Macromolecules, 1995, 28(1):390[10] Mykytiuk J, Armes S P, Billingham N C. Polymer Bulletin,1992,29(1~2):139[11]Pugh C,Pecec V.Polymer Bulletin ,1985,14(2):10910 Hertler W R. Walter R,Sutherlin D M, Ovenall D W, etal. J Am Chem Soc, 1988, 110(17):841[12] Carothers Terrell W ,Mathias Lon, Polym Pre, 1992,33(2):150[13] (1) Kim Y, Choi W, Choi B, et al. Macromolecules,1991, 24: 5006; ( 2 ) Choi W, Kim Y, Choi S.Macromolecules, 1990, 23:5365[14] Sogah D Y, Polym Pre, 1991,32(1):307[15] Sogah D Y, Webster O W. Macromolecules, 1986, 19:1775[16] Liu J K, Wnek G E. Macromolecules 1994,27(15):4080。