谱峰拟合首先确定谱峰拟合的范围
- 格式:pps
- 大小:3.78 MB
- 文档页数:11

内标和外标角度校准角度校准是一种用来校正由仪器和实验的非理想性所引起图谱重大角偏差的手段。
标准参考物质(SRMs)为校准提供峰位置参考。
可以通过菜单“Analyze | Theta Calibration”或者右键单击主工具栏上“theta calibration”按钮调用角度校准对话框。
图8-1给出了两个关于角度校准对话框的例子。
图8-1 使用和不使用PDF重叠的角度校正对话框角度校准分两步:(1)绘制校准曲线;(2)应用校准曲线。
第(1)步要求图谱来源于标准物质,或者包含一个或多个标准物质,这样才能使基准有正确的峰位置。
对话框中列出的SRMs 是从Jade程序文件夹中的jade5.srm文件读入的,用户可以使用文本编辑器浏览和编辑该文件,如果需要可以添加或删除SRM。
如果在缩放窗口中存在一个PDF图谱,Jade将把它作为SRM。
换句话说,在角度校准中PDF图谱取代了内置的SRMs。
如果一个图谱不用于校准,在调用校准之前要移除它。
用户也可以在单张扫描图谱校准中添加数张PDF图谱作为SRMs。
校准曲线是把⊿2θ值作为2θ角的函数进行最小二乘拟合,这里⊿2θ是参考衍射线与待校峰位置之间的差值。
考虑到角偏差的性质和图谱的扫描范围,用户可以选择“Correction Type”以拟合曲线。
例如,如果在校准范围内只有一个标准峰,可以只使用零点偏移校正。
用户可以根据ESD值选择适当的校准曲线,也就是说,可以选择给出小的ESD 值同时丢弃尽量少的数据点的曲线。
如果寻峰没有给出任何峰,Jade将在校准中使用拟合峰列表。
如果寻峰和拟合数据都没有,Jade会在绘制校准曲线之前进行自动寻峰。
要得到最好的结果,就要只使用属于同一SRM相关(relevant)的并且被很好解析的峰。
拟合的Kα1峰适于校准,因为参比峰位置是从使用Kα1波长辐射的参比线的d值计算得来的。
Jade将把参考线与待校峰在指定的2θ误差窗口中匹配。
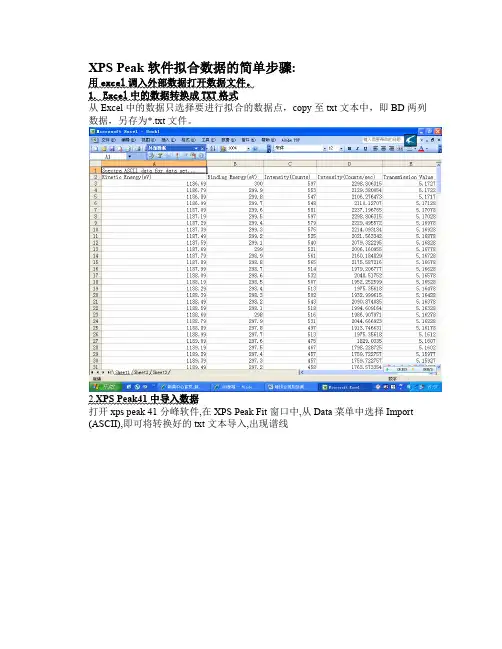
n XPS Peak 软件拟合数据的简单步骤:用excel 调入外部数据打开数据文件。
1. Excel 中的数据转换成TXT 格式从Excel 中的数据只选择要进行拟合的数据点,copy 至txt 文本中,即BD 两列数据,另存为*.txt 文件。
2.XPS Peak41中导入数据打开xps peak 41分峰软件,在XPS Peak Fit 窗口中,从Data 菜单中选择Import (ASCII),即可将转换好的txt 文本导入,出现谱线3.扣背底在打开的Region 1窗口中,点击 Backgrond ,选择Boundary 的默认值,即不改变High BE 和Low BE 的位置,Type 一般选择Shirley 类型扣背底4.加峰选择Add Peak ,选择合适的Peak Type(如s,p,d,f),在Position 处选择希望的峰位,需固定时点fix 前的小方框,同时还可选半峰宽(FWHM )、峰面积等。
各项中的constaints 可用来固定此峰与另一峰的关系。
如W4f 中同一价态的W4f 7/2和W4f 5/2的峰位间距可固定为2.15eV ,峰面积比可固定为4:3等,对于%Lorentzian-Gaussian 选项中的fix 先去掉对勾,点击Accept 完成对该峰的设置。
n 点Delete Peak 可去掉此峰。
再选择Add Peak 可以增加新的峰,如此重复。
注意:% Lorentzian-Gaussian 值最后固定为20%左右。
加峰界面举例:对峰的限制constraints ,峰1的峰位=峰0峰位+1.55.拟合选好所需拟合峰的个数及大致参数后,点XPS Peak Processing 中的Optimise All 进行拟合,观察拟合后总峰与原始峰的重合情况,如不好,可多次点Optimise All合适图谱6.参数查看拟合完成后,分别点XPS Peak Processing窗口总的Region Peaks下方的0、1、2等,可查看每个峰的参数,此时XPS峰中变红的曲线为被选中的峰。

固体核磁分峰拟合1.引言1.1 概述在固体核磁共振(NMR)技术中,分峰拟合是一种常用的分析方法。
当我们对某个固体样品进行核磁共振实验时,通过强磁场作用下,样品原子核会产生特定的共振信号。
这些信号可以被记录并转换为核磁共振谱图,其中包含了丰富的信息。
然而,由于固体样品中原子核的环境复杂,谱图往往呈现多个峰的形式。
为了从谱图中准确提取出各个峰的参数,我们需要借助分峰拟合方法。
分峰拟合可以将谱图中的峰拟合为高斯曲线或者洛伦兹曲线,从而得到每个峰的位置、面积、峰宽等信息。
分峰拟合在固体核磁共振分析中有着广泛的应用。
它可以用于定量分析样品中某个特定分子的含量,还可以帮助我们研究样品的结构和动力学性质。
通过分峰拟合,我们可以得到更加精确和详细的核磁共振谱图信息,为进一步的研究提供重要的基础。
本文将介绍固体核磁共振技术以及核磁共振谱图的分析方法,并重点探讨了分峰拟合的原理和应用。
通过对分峰拟合方法的研究和应用,我们可以更准确地获得样品的核磁共振谱图信息,并为相关领域的研究提供有力支持。
1.2文章结构文章结构部分的内容如下:1.2 文章结构本文主要分为三个部分进行讨论。
首先,在引言部分将对固体核磁分峰拟合的概述进行介绍,包括该技术的基本原理、应用领域以及研究意义。
其次,在正文部分,将详细介绍核磁共振技术的基本知识,包括核磁共振的原理、仪器设备和实验操作方法等内容。
同时,还将探讨核磁共振谱图的分析方法,包括化学位移、耦合常数和峰形分析等方面内容。
最后,在结论部分将详细介绍固体核磁分峰拟合的方法,并讨论其在实际应用中的前景和潜在的发展方向,以及可能的挑战和限制。
通过这样的结构安排,本文旨在给读者提供一个全面而深入的了解固体核磁分峰拟合技术的文章。
1.3 目的本文的目的是探讨固体核磁共振谱图的分峰拟合方法及其在实际应用中的前景。
通过对固体样品进行核磁共振谱图分析,我们可以获取关于样品分子结构、动力学信息以及相互作用的重要信息。

EPR光谱分峰拟合
EPR(电子顺磁共振)光谱是一种常用的分析方法,用于检测物质中的自由基和其他电子自旋态。
EPR光谱通常表现为一个复杂的谱线,由多个峰值组成。
对于EPR光谱的分峰拟合,一般可以通过以下步骤进行:
1. 获取EPR光谱数据:使用EPR仪器获取样品的EPR 光谱数据,并将其导入电脑中进行处理。
2. 基线校正:对EPR光谱进行基线校正,消除背景噪声和其他干扰信号。
3. 分峰拟合:对EPR光谱进行分峰拟合,将其分解成多个峰值。
分峰拟合可以使用各种数学方法,如最小二乘法、高斯拟合等。
4. 分析结果:根据分峰拟合的结果,分析每个峰值对应的自由基类型和浓度,以及其他相关信息。
EPR光谱分峰拟合是一项比较复杂的分析工作,需要具备一定的专业知识和经验。
在实际应用中,通常需要结合其他分析方法和实验手段,以获得更加准确和全面的结果。

拉曼光谱分峰拟合求⾯积(峰强度)教程第⼀步:把记事本中的数据复制(Ctrl+C)第⼆步:打开Origin,点击红框中的第⼀个表格,粘贴(Ctrl+V)3:点击红⾊部分的空⽩部分选中数据。
之后变为⿊⾊4:点击下⽅的红圈中的按钮作图之后出现拉曼光谱图⾸先基线校准。
(有的同学的图是斜着的,所以得基线校准,否则没法求峰⾯积和峰强度)按步骤打开⽅框中的按钮打开对话框选择subtract baseline,之后点上⽅的next下拉菜单,中选择user defined按箭头的提⽰,把add钩去掉,点击Add然后出现基线校准界⾯,点击最曲线最左边的位置,点击键盘上的Enter,⼗字变为圆圈。
之后点击曲线最右边的位置,点击键盘Enter,也可以多点⼏个点让曲线更好看。
之后点击done最后点击箭头处的finish基线校准完成双击图中⽅框中,分别点击横纵坐标,点击scale,横坐标改为800—2000,纵坐标改为0开始,之后就变为下图。
接下⾥,分峰拟合,按照提⽰点击⽅框中的按钮,打开对话框。
选择Gaussian。
点击峰的顶点位置,之后键盘Enter键。
选择另⼀个峰也enter键。
之后左上⾓的fit按钮。
然后yes。
选择右下⾓的project explorer,然后双击中间剪头位置,打开曲线图。
曲线已经拟合好,双击红⾊或绿⾊的线,把line中的线改为3号,为了拟合的曲线更清晰。
有⽊有变的很清晰呢,嘿嘿。
图⽚中有两条拟合曲线,红⾊和绿⾊部分,红⾊是D带,绿⾊是G带。
接下来分别求峰强度(也就是峰⾯积)。
单击红⾊的部分,停顿⼀秒,之后再单击⼀次(注意停顿),选中红⾊曲线。
此时红⾊变为⿊⾊(选中),⽽第⼆峰没有被选中按照提⽰选择计算峰⾯积。
点击下⽅的result log。
⽅框中的area便是峰的强度ID(也就是⾯积)把它复制下来粘贴到Excle中。
同样的⽅法,此时可以点击last use了,求出右边锋的⾯积IG之后把横纵坐标的标签改下,纵坐标可以不要,把⼤框中的内容Delete删掉。

红外光谱分峰拟合
红外光谱分峰拟合是一种分析红外光谱图像的方法,用于确定样品中存在的不同分子或官能团的类型和数量。
下面是红外光谱分峰拟合的基本步骤:
1.数据预处理:将原始红外光谱数据进行预处理,例如进行峰值对齐、基线校正和去噪等处理。
2.初步拟合:使用高斯或洛仑兹函数对谱图进行初步拟合,以确定潜在的谱峰位置。
3.定义拟合函数:根据样品的化学特性和红外光谱数据,选择合适的拟合函数,例如高斯或洛仑兹函数等。
4.拟合参数设置:设置拟合函数的参数,包括峰形、峰位、峰高等参数。
5.拟合优化:通过最小二乘法等方法对拟合函数进行优化,以获得最佳的拟合结果。
6.分析谱峰:对拟合结果进行分析,确定样品中存在的分子或官能团的类型和数量。
7.结果评估:根据分析结果进行结果评估,检查拟合效果和确定的峰位是否准确。
8.红外光谱分峰拟合是一种常用的谱图分析方法,可以帮助科学家们深入研究不同化合物或材料的化学结构和性质,为材料研究、化学分析等领域的应用提供重要的支持。

核磁共振氢谱数据处理核磁共振氢谱是一种非常重要的谱学方法,它可以通过样品中氢原子的化学位移来确定其化学结构。
在实验中,我们首先需要获取氢谱数据,然后对数据进行处理,得到有意义的结果。
本文将介绍关于核磁共振氢谱数据处理的方法。
1.数据预处理在对氢谱数据进行处理之前,我们需要进行一些数据预处理工作,以保证数据的精度和可靠性。
首先,我们需要对谱图进行基线校正,因为在实验中可能会存在噪音、漂移、基线扭曲等干扰,这些因素会影响到数据的准确性。
基线校正方法有多种,其中最为常用的是线性基线校正和曲线匹配法。
在这里,我们以线性基线校正为例,其具体操作方法如下:(1)在谱图中选择一段不包含信号的区域,然后用一条直线连接该区域两端的数据点,以其为基线。
(2)将基线移动到谱图中心位置,然后将谱图按垂直方向上的基线进行切割,得到基线校正后的谱图。
(3)对切割后的谱图进行标度调整,使其最大值等于1。
2.数据解析和谱峰归属在数据预处理完成后,我们可以对氢谱图进行解析,来确定其中每个峰的位置和相对强度,这对于谱峰的归属和分析非常有帮助。
数据解析方法有多种,其中最为常用的是峰积分法和谱峰匹配法。
在这里,我们以峰积分法为例,其具体操作方法如下:(1)首先,将加入TMS作为内标化合物的NMR样品加入到NMR管中,观察样品的NMR谱图。
(2)根据谱图中的化学位移值和相对强度确定每个峰的位置和相对强度。
(3)根据信号的位置和相对强度对每个峰进行归属,可以通过与已知样品的谱图比对来实现。
3.分析谱图在谱峰归属之后,我们就可以对氢谱图进行进一步分析了,以便确定样品的化学结构和分子式。
常见的分析方法有多种,下面列举了其中的一些:(1) 根据谱峰的位置和相对强度绘制化学位移图谱,并与已知化合物的谱图进行比对,以确定样品的结构。
(2) 根据谱峰之间的耦合相互关系,构建耦合常数矩阵,以确定氢原子之间的相对位置和空间关系。
(3) 根据氢谱图中的峰形变化,确定样品的对称性和结构特征。

碳材料拉曼分峰拟合
碳材料拉曼分峰拟合是一种常用的方法,用于分析和表征碳材料的结构和物理性质。
拉曼分光光谱是一种非常敏感的分析技术,能够探测和分析材料的微观结构和振动特性。
拉曼光谱图可以通过拟合各种谱峰来解析材料的结构和化学组成。
在碳材料的拉曼光谱中,常见的谱峰有D带、G带和2D带,它们分别对应于材料的结构和振动特性。
通过拉曼分峰拟合,可以准确地确定这些谱峰的位置、形状和强度,并进一步分析材料的结构和性质。
此外,拉曼分峰拟合还可以用于研究碳材料的缺陷、应力和表面性质等方面,具有重要的应用价值。
- 1 -。

XPS谱峰拟合注意事项XPS(X-ray Photoelectron Spectroscopy)是一种常用的表面分析技术,通过测量材料中的光电子能谱来获取元素的化学状态和表面化学组成信息。
在进行XPS谱峰拟合时,需要注意以下几个方面:1.谱峰选择:首先需要选择合适的XPS谱峰进行拟合。
对于元素的3d谱峰,一般选用高分辨率谱进行拟合;而对于元素的2p谱峰,通常选用低分辨率谱拟合。
此外,还需要根据样品的化学组成选择合适的XPS谱峰。
2.光电子能谱线宽:在进行谱峰拟合之前,需要确定光电子能谱的线宽。
光电子能谱的线宽包括仪器效应和样品表面等原因引起的线宽。
通常情况下,可以通过测量标准物质的XPS谱线来确定仪器效应的线宽。
如果样品表面存在不均匀性,还需要考虑表面扩散等因素。
3. 谱峰形状:在进行XPS谱峰拟合时,需要选择合适的谱峰形状函数。
常用的谱峰形状函数有高斯函数、洛伦兹函数和Voigt函数等。
对于复杂的谱峰,可以采用多个形状函数的组合进行拟合。
4.背景拟合:在XPS谱峰拟合中,背景信号会对谱峰的拟合结果产生影响。
背景信号包括激光连续谱、底部散射等引起的背景。
一般情况下,可以采用多项式函数进行背景拟合。
而对于特殊样品,可能需要使用更复杂的数学函数进行背景拟合。
5.谱线强度校正:XPS谱线的强度受到多种因素的影响,如分析深度、探针电强度、距离等。
在进行谱峰拟合时,需要对谱线的强度进行校正,以获得准确的表面化学组成信息。
6.谱峰重叠处理:在一些情况下,不同元素的谱峰可能会发生重叠。
为了准确区分不同元素的贡献,需要进行谱峰重叠处理。
常见的处理方法包括利用化学键能的差异、XPS波长的差异等。
7.数据处理与误差分析:在进行XPS谱峰拟合之后,还需要对数据进行处理和误差分析。
可以计算每个元素的相对百分含量、角分辨率、信噪比等指标。
此外,还需要对拟合结果进行误差分析,以评估数据的可靠性和拟合的准确性。
总之,进行XPS谱峰拟合需要综合考虑实验条件、谱峰形状、背景拟合等多种因素。

核磁数据处理方法标题:核磁数据处理方法引言概述:核磁共振(NMR)是一种常用的分析技术,广泛应用于化学、生物、医学等领域。
在核磁共振实验中,数据处理是非常重要的环节,能够帮助研究人员从海量数据中提取有用信息。
本文将介绍核磁数据处理的方法和技巧。
一、数据预处理1.1 去噪处理:在核磁共振实验中,由于仪器本身的噪声和样品的环境因素等原因,数据中可能存在噪声。
去噪处理可以帮助提高数据质量,减少误差。
1.2 相位校正:相位校正是核磁数据处理中的一项重要步骤,可以帮助消除数据中的相位差异,提高信噪比。
1.3 基线校正:基线漂移是核磁数据中常见的问题,通过基线校正可以减少基线漂移对数据分析的影响。
二、谱图处理2.1 傅立叶变换:核磁共振数据通常是在时域上采集的,通过进行傅立叶变换可以将时域数据转换为频域数据,得到核磁共振谱图。
2.2 化学位移标定:在核磁共振谱图中,化学位移是一项重要的参数,可以通过标定化学位移来确定样品中不同原子的位置。
2.3 峰识别和积分:对核磁共振谱图进行峰识别和积分可以帮助确定样品中不同化合物的含量和结构。
三、数据分析3.1 谱峰拟合:对核磁共振谱图中的峰进行拟合可以帮助确定峰形参数,如峰高、峰面积等,进而得到样品的定量信息。
3.2 化学结构推断:通过核磁共振数据的分析,可以推断样品的化学结构,帮助研究人员了解样品的组成和性质。
3.3 数据挖掘:利用机器学习和数据挖掘技术,可以对核磁共振数据进行深入分析,挖掘隐藏在数据中的规律和信息。
四、谱图解释4.1 化学位移解释:化学位移是核磁共振谱图中的重要参数,可以通过化学位移的解释来确定样品中不同原子的化学环境。
4.2 耦合常数分析:耦合常数是核磁共振谱图中的另一个重要参数,可以通过耦合常数的分析来推断分子内的相互作用关系。
4.3 结构确认:通过对核磁共振谱图的解释和分析,可以确认样品的化学结构,为后续研究提供重要参考。
五、数据可视化5.1 二维谱图绘制:二维核磁共振谱图是一种常用的数据可视化方式,可以直观地展示样品中不同原子之间的相互作用关系。

XPS谱峰拟合注意事项(一)X光电子能谱分析的基本原理X光电子能谱分析的基本原理:一定能量的X光照射到样品表面,和待测物质发生作用,可以使待测物质原子中的电子脱离原子成为自由电子。
该过程可用下式表示:h n=E k+E b+E r(1)其中:h n:X光子的能量;E k:光电子的能量;E b:电子的结合能;E r:原子的反冲能量。
其中Er很小,可以忽略。
对于固体样品,计算结合能的参考点不是选真空中的静止电子,而是选用费米能级,由内层电子跃迁到费米能级消耗的能量为结合能E b,由费米能级进入真空成为自由电子所需的能量为功函数Φ,剩余的能量成为自由电子的动能E k,式(1)又可表示为:hn=Ek+Eb+Φ(2)Eb=hn-Ek-Φ(3)仪器材料的功函数Φ是一个定值,约为4eV,入射X光子能量已知,这样,如果测出电子的动能Ek,便可得到固体样品电子的结合能。
各种原子,分子的轨道电子结合能是一定的。
因此,通过对样品产生的光子能量的测定,就可以了解样品中元素的组成。
元素所处的化学环境不同,其结合能会有微小的差别,这种由化学环境不同引起的结合能的微小差别叫化学位移,由化学位移的大小可以确定元素所处的状态。
例如某元素失去电子成为正离子后,其结合能会增加,如果得到电子成为负离子,则结合能会降低。
因此,利用化学位移值可以分析元素的化合价和存在形式。
(二)电子能谱法的特点(1)可以分析除H和He以外的所有元素;可以直接测定来自样品单个能级光电发射电子的能量分布,且直接得到电子能级结构的信息。
(2)从能量范围看,如果把红外光谱提供的信息称之为“分子指纹”,那么电子能谱提供的信息可称作“原子指纹”。
它提供有关化学键方面的信息,即直接测量价层电子及内层电子轨道能级。
而相邻元素的同种能级的谱线相隔较远,相互干扰少,元素定性的标识性强。
(3)是一种无损分析。
(4)是一种高灵敏超微量表面分析技术。
分析所需试样约10-8g即可,绝对灵敏度高达10-18g,样品分析深度约2nm。
X射线光电子能谱中的分峰处理孙博文;余红雨;钱东金;陈萌【摘要】文章详细介绍了X射线光电子能谱分峰操作的流程,对谱图中单峰和多峰的荷电位移、数据平滑、背景扣除、谱峰确认和多高斯拟合操作的数学流程进行了图解.我们认为,具有足够连续宽度和强度的峰值处才可以被判定为峰;谱图中可以进行含量定量的谱峰强度应在背景涨落的10倍以上;最后得到的分峰结果,还应进行统计检验.【期刊名称】《大学化学》【年(卷),期】2017(032)008【总页数】7页(P53-59)【关键词】背景扣除;谱峰指认;谱峰拟合;统计检验【作者】孙博文;余红雨;钱东金;陈萌【作者单位】复旦大学化学系,上海200433;复旦大学化学系,上海200433;复旦大学化学系,上海200433;复旦大学材料系,上海200433【正文语种】中文【中图分类】G64;O6X射线光电子能谱(XPS)是使用X射线(约1000-1500 eV)激发样品表面,通过测量激发出的光电子能量分布而确定材料的表面组成和电子结构的。
由于XPS对样品损伤小且灵敏度高,因而在表面层结构分析与界面物化性质测定方面有着广泛的应用。
在实际科研工作中,XPS在研究材料表面性质、分析元素组成、测定半导体价带等方面都有着广泛的应用,对于从事无机化学、物理化学、分析化学等领域的科研人员来说都是一种强有力的手段。
XPS中,谱峰的化学位移与材料的化学结构和原子价态有关。
相同激发源及谱仪接收条件下,光电子峰的面积大小也可以表征该信号中的元素含量。
因此,XPS表征技术对于化学材料的表面研究来说必不可少。
但目前利用相关软件对XPS分峰时,在确定谱峰数量、谱峰位置时都有一定随意性,这与科研工作的严谨性相悖。
因此,本文主要讨论如何借助规范的数学手段确定XPS谱峰的准确位置,并给出峰面积。
本文从荷电校正、谱图平滑、谱峰指认、曲线拟合、统计检验等方面对分峰过程进行了介绍。
处理数据时所使用的软件主要为Origin 8.0和XPS Peak 4.2。
XPS拟合峰的标准包括以下步骤:
1. 峰形选择:根据实际谱图的特点选择合适的峰形函数,如高斯函数、洛伦兹函数和泊松函数等。
2. 背景拟合:在进行峰拟合之前,需要进行背景拟合,将谱图中的背景信号剔除,以提高峰拟合的准确性。
背景拟合可以选择线性函数、二次函数或指数函数等进行拟合,具体选择应根据实验数据的特点而定。
3. 峰位选择:根据实验谱图中峰的形状和位置合理选择。
一般情况下,峰位应选择在实验峰的中间位置,避免选择在峰的边缘位置,以减小拟合误差。
4. 拟合范围:拟合范围应包括峰的整个轮廓,但避免包含其他峰的信息。
5. 拟合参数:拟合参数需要根据实验数据的特点进行调整,以保证拟合的准确性。
6. 峰的重叠问题:当样品中存在多个元素时,它们的的光电子峰可能会重叠在一起,导致峰的形状复杂。
这时可以采用多峰拟合的方法,将重叠的峰分解为多个峰,从而得到更准确的峰参数。
请注意,以上步骤只是一种常用的数据处理方法,具体操作时可能需要根据实际情况进行调整。
如果想要深入了解,可以请教物理或化学领域专业人士。
XPS Peak 软件拟合数据的简单步骤用excel 调入外部数据打开数据文件。
1. Excel 中的数据转换成TXT 格式 从Excel 中的数据只选择要进行拟合的数据点, copy 至txt 文本中,即BD 两列 数据,另存为*.txt 文件。
1 JJk □ llETfrjOft K±C«! 1 •加x - 1 !■X] nn? MA u WtUT'舶人© teStliJ 3(p rfc-1) 41L1Q伍旦!UP 琴..wum"闘 I2 * *RB .C __________ I __________ l 艮H 1 1 Ijc-r :' ■ Ti LL-:1. |J-31. -3 f o~ dit- LT 1,,..2 Kint^Lc EneruyD ■"indlj ■■& Kne-好 rV [nirt^liy (-\njrn.:.:s ]n- r.n :i ty (讥〕un 」.:=:/ 土」 Tf .instilltF -.ni血3 118^693训2前乩 PO-315 5.1T2T■; 1136. ?9 阿,9 2129.泊加E 1136.対 EM.S2106. 27K4735.1T17 G1:S6.鹃 2W.?211I&. 127CT5.1112S Tusr. os 附K 531 325T. 1 州T 隔 5.1T0T& 8 11S7.19 ZM.B 彌229B. BW3LB 5.1T02E I '3 1187.235792229.495572 5-K97E w 1167. 392?3.35T5 2ZL4. D93134 5.155Z3 11 11S7,刖5252QEL5&334Z 5UW7E12 lLS'r. 59 2OTul M0 2Cl9L 32二2涙 5L 1SE2E 13 ns?. o9 咖B21 ... .uB. ] toyti. 5.167'I ': 14 1187.795C12160. 184E21 B L 16I2B沁£F K 1T 1S Fd i 21 iss.:gS07 1<S2. 2號5溯ilSS.対2^.4 513 l^TE. 35614UM ■ 2垃』5U2140蛊痢运 5.1E t2Ei 「-1讥厂 P I 花E 茨£ 1託匸 21T 反 587Z16ISTTS. 206T7T204E. 5175223S r 6 11S7.旳 11E7.轴 11SS- 0921 1183.49 2^2W3ZZ , iiaa.592^1 513 23 1138. 69 298516 24 1188. 79 2<9BS125 1138-的筑1188,^2W.7S19 27 . Q8109溯」*?5US9-13 Z9T.511 熬 2'3 Z97.4知11 S3.33 洌;311LE3-的2SL2H « * *1 \汕“Li /加彎U //1<2u.^. r.[J8S5. we伽、&吟1钠5.16328 1S86. 9&T9715.1S2T8 2沁E •隔923 S, 1SE28 1918.746631 5.1517E :157^.35618>.H1.18? 9. '335 5.16071T9B> ZZ37ZS 5.UQZ ITS 乳 7ZZF5T 5,15977ITS?. 7227&T MW1WJ ST33M 4EE/m□I'EfS二.F 宿 島(S 叶 ■八討置嘗住口英用 C 1 秤ft 耳]▼ firii-i 3E] l..j..U h bnl..• m 百 7 j * ll :V2.XPS Peak41中导入数据 打开xps peak 41分峰软件,在XPS Peak Fit 窗口中,从Data 菜单中选择Import(ASCII),即可将转换好的txt 文本导入,出现谱线:涵3M 晦梓" 溯0blarthni fwniy» *V'l3扌口背底在打开的Region 1窗口中,点击Backgrond,选择Boundary的默认值,即不改变High BE 和Low BE的位置,Type —般选择Shirley类型扣背底4加峰选择Add Peak,选择合适的Peak Type如s,p,d,f),在Position处选择希望的峰位,需固定时点fix前的小方框,同时还可选半峰宽(FWHM )、峰面积等。
XPS谱峰拟合注意事项XPS谱峰拟合注意事项(⼀)X光电⼦能谱分析的基本原理X光电⼦能谱分析的基本原理:⼀定能量的X光照射到样品表⾯,和待测物质发⽣作⽤,可以使待测物质原⼦中的电⼦脱离原⼦成为⾃由电⼦。
该过程可⽤下式表⽰:h n=E k+E b+E r (1)其中:h n:X光⼦的能量;E k:光电⼦的能量;E b:电⼦的结合能;E r:原⼦的反冲能量。
其中Er很⼩,可以忽略。
对于固体样品,计算结合能的参考点不是选真空中的静⽌电⼦,⽽是选⽤费⽶能级,由内层电⼦跃迁到费⽶能级消耗的能量为结合能E b,由费⽶能级进⼊真空成为⾃由电⼦所需的能量为功函数Φ,剩余的能量成为⾃由电⼦的动能E k,式(1)⼜可表⽰为:hn=Ek+Eb+Φ(2)Eb=hn-Ek-Φ(3)仪器材料的功函数Φ是⼀个定值,约为4eV,⼊射X光⼦能量已知,这样,如果测出电⼦的动能Ek,便可得到固体样品电⼦的结合能。
各种原⼦,分⼦的轨道电⼦结合能是⼀定的。
因此,通过对样品产⽣的光⼦能量的测定,就可以了解样品中元素的组成。
元素所处的化学环境不同,其结合能会有微⼩的差别,这种由化学环境不同引起的结合能的微⼩差别叫化学位移,由化学位移的⼤⼩可以确定元素所处的状态。
例如某元素失去电⼦成为正离⼦后,其结合能会增加,如果得到电⼦成为负离⼦,则结合能会降低。
因此,利⽤化学位移值可以分析元素的化合价和存在形式。
(⼆)电⼦能谱法的特点(1)可以分析除H和He以外的所有元素;可以直接测定来⾃样品单个能级光电发射电⼦的能量分布,且直接得到电⼦能级结构的信息。
(2)从能量范围看,如果把红外光谱提供的信息称之为“分⼦指纹”,那么电⼦能谱提供的信息可称作“原⼦指纹”。
它提供有关化学键⽅⾯的信息,即直接测量价层电⼦及内层电⼦轨道能级。
⽽相邻元素的同种能级的谱线相隔较远,相互⼲扰少,元素定性的标识性强。
(3)是⼀种⽆损分析。
(4)是⼀种⾼灵敏超微量表⾯分析技术。
分析所需试样约10-8g即可,绝对灵敏度⾼达10-18g,样品分析深度约2nm。
核磁数据处理方法核磁共振(NMR)是一种常用的分析技术,广泛应用于化学、生物、医学等领域。
在核磁共振实验中,我们通常会获得一系列的核磁共振谱图,这些谱图包含了样品中各种化合物的信息。
为了准确地分析和解释这些数据,需要进行核磁数据处理。
核磁数据处理的目标是提取和分析核磁共振谱图中的信息,以便确定样品中的化合物类型、结构和相对含量等。
下面将介绍一些常用的核磁数据处理方法。
1.基线校正核磁共振谱图中往往存在基线漂移的问题,即谱图的底部不平整。
基线校正是一种常用的预处理方法,旨在消除基线漂移的影响。
常见的基线校正方法包括多项式拟合、直线拟合和基线修正等。
2.化学位移校正化学位移是核磁共振谱图中的一个重要参数,用于确定样品中的化合物类型。
然而,由于仪器和实验条件的不同,不同样品的化学位移值可能存在一定的偏差。
因此,需要进行化学位移校正,以便准确地确定化学位移值。
化学位移校正往往使用内部标准物质进行,通过与内部标准物质的化学位移进行比较,确定样品中各化合物的化学位移值。
3.谱峰拟合核磁共振谱图中的谱峰代表了不同核的共振信号,通过谱峰的位置、形状和强度可以确定样品中的化合物结构和相对含量。
谱峰拟合是一种常用的谱峰分析方法,通过对谱峰进行数学拟合,可以准确地确定谱峰的位置、宽度和强度等参数。
4.峰面积计算谱峰的面积与核磁共振信号的强度成正比,可以用于确定样品中不同化合物的相对含量。
峰面积计算是一种常用的定量分析方法,通过对谱峰的面积进行测量和计算,可以确定样品中各化合物的相对含量。
5.谱图解析谱图解析是核磁数据处理的最终目标,通过对核磁共振谱图中的各峰进行解析和分析,可以确定样品中的化合物结构和相对含量。
谱图解析需要结合化学知识和谱图解析软件,通过对谱峰的位置、形状和强度等参数进行分析,可以确定样品中各化合物的结构和相对含量。
总结:核磁数据处理是核磁共振实验中的重要环节,通过对核磁共振谱图进行基线校正、化学位移校正、谱峰拟合、峰面积计算和谱图解析等处理方法,可以准确地提取和分析核磁共振谱图中的信息,确定样品中的化合物类型、结构和相对含量等。
第七章数据处理方法一、谱图分析二、谱图修正三、谱峰拟合四、数值方法(NLLSF与TFA)四、数据处理方法●数据处理包括:●谱图分析●谱图修正●谱峰拟合●数值方法(NLLSF与TFA)一、谱图分析●鉴别谱峰(自动/手动按照能量范围或元素)●定性分析(元素鉴别、化学态分析)●定量分析(原子浓度、峰面积、归一化峰面积、传输函数修正、灵敏度因子)鉴别谱峰(按元素)鉴别谱峰(按能量范围)二、谱图修正●数据平滑:Savitzky-Golay, 傅立叶滤波●本底去除:线性, Shirley, Tougaard●微分与积分●校正荷电位移●谱图比较/覆盖, 归一化谱图1、数据平滑●实验谱图中包含有测量随机噪声。
●数据平滑的目的就是这些高频噪声成分而不失真地保留原始谱图中包含的涉及峰高和峰型的所有信息。
提高谱线的信噪比。
●存在两种基本的平滑方法:使用卷积平滑函数与应用傅里叶分析的频率滤波。
●常用第一种平滑方法,尤其是多点移动平滑。
●最常用的卷积程序是三次/四次函数—Savitzky-Golay,其次可采用高斯函数。
●当平滑点数取谱图中可分辨的最窄峰的FWHM所含的数据点数时,Savitzky-Golay函数效果最佳,失真低。
2、本底去除●在XPS谱和AES直接谱中,通常为较小的谱峰叠加在大的本地之上。
如果要检查谱峰的细节,在某些情况下就需要进行本底去除(如定量时测量谱峰强度时)。
●最简单的本底去除方法是在用户感兴趣的谱峰两端指定点间作直线—线性。
●线性本底通常误差较大,是非物理的。
●线性本底的改进涉及到的物理真实逼近—Shirly本底。
线性本底非线性本底-Shirley Method ⎥⎦⎤⎢⎣⎡+-=∑=)(5.0)(k x k x i i y y y h Q ●使用最普遍的非线性背景扣除方法●该方法认为能量损失是常数, 谱线上任一点由非弹性散射电子引起的背景, 只来源于更高动能电子的散射, 正比于更高动能的积分光电子强度(面积)●所以任一能量的本底都正比于光电子能谱中具有较高能量电子的总数目。
●B(x) = b+ ( a-b)Q/(P + Q)式中:P + Q 为扣除背景后峰的总面积;Q 为动能E 以上的光电子的积分强度●因为背景B(x)是未知的待求量,开始无法计算面积P 和Q, 为此首先用常数背景B1 作为初值, 计算出P 、Q 后再计算出新的背景, 如B2, 如此反复迭代, 直至收敛为止。
Smart backgrounShirley backgroundSmart backgroundSmart 本底源自于Shirley 本底,但反复调整本底位置使得本底不跑到数据曲线之上。
这尤其适用于有较宽能量范围的双线谱峰定量。
3、微分谱(Derivative Spectrum)●微分谱提供了一种在某些情况下简单快速地确定峰位的有用方法。
●二次微分谱的负峰位近似对应于原始谱中重叠峰的位置。
4、荷电校正方法在实际的XPS分析中,一般采用内标法进行校准。
即在实验条件下,根据试样表面吸附或沉积元素谱线的结合能,测出表面荷电电势,然后确定其它元素的结合能。
●外来污染碳氢化合物参考●用真空系统中最常见的有机污染碳的C 1s的结合能作为参考,常用284.6 eV至285eV作为参考结合能。
以测得的C1s谱线的结合能与参考值之差为荷电校正值 ,来校正谱图中其它谱峰的测量结合能值。
●尽管外来污染碳作静电荷电校正存在局限性和不确定性,但它仍是最方便和最常用的技术。
“污染碳”的C 1s结合能比较C (1s) BE of Hydrocarbons C (1s) BE of Hydrocarbons元素自然氧化物离子刻蚀金属BE差元素自然氧化物离子刻蚀金属BE差Ag285.5284.70.8Nb285.1284.90.2Al286.3285.1 1.2Ni285.4284.90.5As284.6284.7-0.1Pb285.6285.20.4[FO] B284.6285.2-0.6Pd285.3284.2 1.1Be285.6284.4 1.2Re284.5285.0-0.5Bi285.4284.80.6Rh284.6284.0*0.6Cd286.0285.01Sb285.0284.40.6[FO] Co285.5284.4 1.1Sc285.9286.8*-0.9Cr285.1284.90.2Se284.3*284.20.1Cu284.7284.7±0.0Si285.7284.90.8Fe285.2284.40.8Sn285.2284.80.4Ga286.1285.60.5Ta284.8284.60.2Ge285.7284.5 1.2Te284.8284.20.6Hf286.2286.10.1Tl285.4285.20.2[FO] In285.4284.90.5V285.1285.2-0.1[FO] Ir285.4285.4±0.0W285.0285.1-0.1Mg286.5284.4 2.1[FO]Y286.7286.7±0.0[FO] Mn284.8286.3-1.5Zn285.8284.90.9Mo284.8285.2-0.4Zr285.9285.40.5荷电校正方法●內标参考●利用样品材料中已知状态元素的结合能值作为参考值进行荷电校准。
如用碳校正,即为碳內标。
●衬底参考●涉及导电衬底上的薄膜研究工作,常以导电衬底元素的结合能作为绝缘覆盖层材料的参考。
●注入惰性气体●向样品注入Ar作内标物有良好的效果。
Ar具有极好的化学稳定性,适合于溅射后和深度剖面分析,且操作简便易行。
三、谱峰拟合●峰拟合可使彼此靠近重叠的峰分别精确测定并且能够更精确地定量。
同时也能够对具有化学位移的元素不同化学态进行准确的分辨和定量。
●可对多个相互重叠峰的谱峰拟合●谱峰拟合参数:峰位中心, 宽度, 高度, 形状L/G比等●不对称拖尾参数(混合, 指数, 高度)●交互式; 添加/移动/修改/删除峰●固定、关联或约束峰参数●快速、通用和交互式的谱峰拟合工具是数据处理软件的一项重要组成部分●可控制谱峰的数目、峰位置、峰宽度、罗仑兹/高斯(L/G) 比m 。
本底类型由用户来选择(线性、Shirley 或Smart)。
●可进行峰参数的关联和固定,并且不对称峰需要时也可以拟合。
●谱峰拟合函数使用高斯-洛伦兹(Gaussian –Lorentzian )乘积函数:220220exp[(1)ln 2()/]()1()/m x x f x H m x x ββ--⋅⋅-=⋅+⋅-这里,H =峰高,β=FWHM/2, m =L/G 混合比拟合方法:•首先确定谱峰拟合的范围(标出起点和终点位置)在峰拟合模式状态下根据需要添加新峰到峰拟合表中。
峰参数可修改谱峰拟合时的考量●谱峰拟合时应注意和考虑:●子峰的个数依赖于峰形和可能的化学环境,各子峰应有合理的物理意义;●合理的半高宽(FWHM) , 一般主峰0.8 ~1.8 eV;●合理的L/G比,一般0.25 ~0.35,默认值:0.3;●对双峰还需特别考虑:两个峰间的合理间距和强度比(可进行关联、固定和约束)两峰间的裂距大小可在XPS手册中查到两峰的面积比一般为:2p:2p3/2 = 1 :21/2:3d5/2 = 2 :33d3/24f5/2 :4f7/2 = 3 :4●过渡金属元素(Fe, Co, Ni)2p峰不对称性较大,拟合较困难聚合物C 1s拟合例Asymmetry of Fe © 2007 XPS International LLCAsymmetry of Fe2O3© 2007 XPS International LLCAsymmetry of Co © 2007 XPS International LLCAsymmetry of CoO © 2007 XPS International LLCAsymmetry of Ni © 2007 XPS International LLCAsymmetry of NiO © 2007 XPS International LLC不对称性峰参数的影响●对于简单不对称峰型谱可引入拖尾函数来拟合●Y=H[GL+(1-GL)*T]●拖尾函数T=TM*CTH+(1-TM)Exp(-DX*ET)其中:CTH=常数尾高比;(1=尾高与峰高相等)TM=尾混合比;(0=指数尾,1=常数尾)ET=指数尾比;(1=无拖尾,典型值: 0.03~0.1)DX =离开峰中心的距离四、数值方法非线性和线性最小二乘法拟合,目标因子分析可应用于包括深度剖析、线扫描和图象在内的多轴数据。
这些拟合方法使得元素或化学物种的重叠得以分离。
1、目标因子分析(TFA)●当数据中包含有多维谱时(深度剖析、线扫描、角分辨XPS等),通常适合于组合使用目标因子分析(TFA)和最小二乘法拟合。
用这种方法,谱中细微的差别可用于测定数据集中不同化学态变化的情形。
●某些纯金属峰并非是简单的高斯-洛仑兹峰型,而是难以拟合的不对称峰型。
●TFA假定剖析数据由共同的谱元的线性组合构成。
TFA仅能确定谱元的数目但并不能鉴别它们。
●元谱可由TFA定义。
比如SiO2/Si的深度剖析,氧化硅的参考谱取自氧化物层中,而元素硅的参考谱取自溅射后衬底中的某一级谱。
原理方法(Procedure)●TFA选取出剖析中最有意义的层次(level),剖析数据中的每个层次都用这些元谱的线性或非线性组合来进行拟合。
●然后计算每个层次的吻合度,并与用户定义的信噪比限值比较。
若此一计算值大于定义的信噪比限值,就认为拟合是满意的。
●若任一层次不能满意地拟合,则该层次增添作为元谱,然后每一层次都以此两谱来进行拟合。
继续这一过程来增添额外的元谱直到所有层次都拟合到比信噪比限值更好时为止。
必需的谱数即为剖析中有意义的元谱数。
注意作为元谱的这些谱可能实际上不包含纯参考谱,因而其强度剖析可能会是负值。
每个层次元谱的线性组合产生表观剖析谱。
●当TFA确定了主元后,它自动用这些元谱执行数据的线性最小二乘法拟合。
2、非线性最小二乘法拟合(NLLSF)●在许多方面NLLSF谱峰拟合与TFA的扩充。
优点是真实峰型可应用于数据集中。
TFA仅支持真实峰型的线性组合,所以分析中移动的峰就不能精确测定。
●在某些实验中,数据中存在有能量位移。
在这种情况下可以使用NLLSF,因此荷电位移峰并不作为不同的化学态来处理。
●NLLSF假定剖析数据由共同的元谱的线性组合构成但其峰位在深度剖析中可以变化(如荷电造成)。
NLLSF对话框点击打开NLLSF对话框定义参考元谱参考元谱可从不同的来源获取●参考元谱位于多级数据集中:●最简单的方法是使用来自于多级数据集中的参考谱。