Neon电转染操作步骤
- 格式:doc
- 大小:19.00 KB
- 文档页数:2

细胞电转染原理
细胞电转染是一种常用的细胞遗传转化方法,利用外加电场介导DNA或其他分子的传递到细胞内。
其原理可以概括为以下几个步骤:
制备DNA或其他分子:首先需要制备目标DNA或其他分子,例如质粒DNA、siRNA、蛋白质等。
细胞预处理:将目标细胞进行预处理,通常包括细胞培养、收获、洗涤等步骤。
预处理的目的是使细胞在转染过程中更容易接受外源DNA或其他分子。
细胞电转染:将目标DNA或其他分子与转染试剂混合,并将混合物加入转染电池中,其中包含了目标细胞。
转染电池中的电极会产生一个外加电场,电场的方向和强度可以调节。
外加电场的作用是在细胞膜上形成暂时的孔隙,使外源DNA或其他分子能够进入细胞内。
恢复细胞状态:在电转染后,细胞需要一段时间来恢复并允许外源DNA或其他分子在细胞内进行表达或发挥功能。
这个过程可能涉及到细胞膜修复和内部信号传导的重建。
细胞电转染的效率受到多种因素的影响,包括电场强度、脉冲持续时间、细胞类型和转染试剂的选择等。
通过优化这些参数,可以提高细胞电转染的效率和成功率。

细胞转染步骤细胞转染是一种将外来脱氧核糖核酸(DNA)或核苷酸序列引入细胞内的技术。
这个过程可以用于研究基因功能、病毒感染和基因治疗。
在细胞转染过程中,有几个重要步骤需要注意。
下面是一个关于细胞转染步骤的简要介绍。
第1步:提取DNA或RNA在细胞转染前,需要先准备好DNA或RNA。
其中,DNA可通过PCR扩增、酵母合成,或从细胞中提取。
RNA则可通过反转录酶聚合酶链式反应(RT-PCR)反转录,然后进行DNA扩增。
第2步:选择转染方法目前有很多种不同的细胞转染方法,包括电穿孔、热休克、化学转染等。
要选择适合的转染方法,需要考虑许多因素,如细胞类型、转染效率和毒性等。
常用的转染剂有聚乙烯醇、脂质体、钙磷酸盐等。
第3步:制备转染复合物转染复合物是转染时添加的液体溶液,用于将DNA或RNA引入细胞。
制备复合物通常需要将DNA或RNA与转染剂混合,然后使其与乙醇或其他化学物质形成稳定的颗粒,最终形成复合物。
转染时需要将制备好的转染复合物直接加入培养基中,让细胞与其接触。
接触后,DNA或RNA会被吸附到细胞表面并被内吞到细胞内部。
转染过程通常需要一定程度的时间,具体时间取决于细胞类型和转染复合物的选用。
第5步:培养和维护转染成功后需要对细胞进行培养和维护。
这通常需要注意以下几个因素:1)细胞浸润度:细胞密度应该适合,以便观察细胞形态和产物的表达量。
2)培养基成分:为了保持细胞生长,需要选择适当的培养基成分。
3)药物筛选:转染后可能需要添加药物对细胞进行筛选,以便筛选出目标基因。
4)稳定性维护:为了保证目标基因稳定的表达,需要适当地维护和稳定细胞系。
以上是细胞转染过程中的几个关键步骤。
在进行细胞转染时,需要注意步骤的顺序和方法的选择,以便提高转染效率和成功率。

脂质体转染原理及操作步骤脂质体转染是一种常用的基因传递技术,用于将外源的核酸(例如DNA、RNA)转入细胞内。
脂质体是由人工合成的类似于细胞膜的双层脂质膜组成,具有较好的生物相容性和可降解性,因此可以用来包裹并传递外源核酸进入目标细胞内。
本文将详细介绍脂质体转染的原理和操作步骤。
脂质体由两个疏水性脂质尾部和一个亲水性脂质头部组成,形成类似生物细胞膜的双层结构。
这种双层结构使得脂质体可以包裹水溶性的外源核酸,并形成脂质体-核酸复合物。
当复合物与细胞膜接触时,由于脂质体的类似性,这些复合物可以与细胞膜融合,并释放外源核酸进入细胞。
然而,简单地将脂质体-核酸复合物添加到细胞培养基中并不能有效地将外源核酸转入细胞内。
为了提高转染效率,常常需要使用电穿孔或钙磷共沉淀等方法,以增加细胞膜的通透性。
操作步骤:1.准备工作:-完整的脂质体转染试剂盒。
-DNA或RNA溶液。
-靶细胞。
-无菌操作条件下的实验室。
2.重组脂质体的合成:-按照试剂盒说明将脂质体复合物合成。
-使用无菌的工具和试剂。
3.核酸引物的制备:-将外源DNA或RNA与引物混合,使其形成核酸复合物。
4.细胞处理:-将培养皿中的细胞进行冲洗,去除培养基。
-将适量的细胞培养基添加到培养皿中,以保证细胞的正常生长。
5.导入核酸:-将所制备的核酸复合物滴加到细胞培养基中。
-将细胞培养皿置于无菌环境中,以便细胞吸收和利用核酸。
6.细胞培养:-按照细胞类型和所使用的培养基,将细胞置于恰当的培养条件下,并进行培养。
-根据需求添加所需要的生长因子和抗生素。
7.观察和分析:-在一定的培养时间后,观察细胞是否发生了显著的变化。
- 使用适当的实验方法(如PCR、Western blot等)检测外源基因的表达。
-观察并记录实验结果。
以上是脂质体转染的原理和操作步骤。
脂质体转染是目前常用的基因传递技术之一,可以用于研究基因功能、疾病治疗等领域。
随着技术的不断发展,转染效率和特异性将逐渐提高,为研究和治疗带来更多的机会和挑战。

细胞转染操作方法一、细胞转染简介细胞转染是指将外源性DNA、RNA、蛋白质等分子通过物理或化学手段导入到细胞内,从而改变细胞的基因表达或功能。
常见的细胞转染方法包括病毒载体介导转染、电穿孔法、钙磷共沉淀法和化学转染法等。
其中,化学转染法是最为常用的方法之一,因为它具有操作简便、成本低廉和适用于多种类型的细胞等优点。
二、实验前准备1. 细胞培养:选择适当的培养基和培养条件,使得细胞处于生长状态。
2. 转染试剂:选择合适的化学转染试剂,如Lipofectamine 2000、PolyJet等。
3. 质粒DNA:准备所需的质粒DNA,并进行纯化和测定浓度。
4. 培养皿:准备适当大小和形状的培养皿,以满足实验需要。
5. 显微镜:准备显微镜以观察细胞转染效果。
三、实验步骤1. 细胞接种:将需要转染的细胞接种到培养皿中,使其达到适当的生长状态。
2. 质粒DNA和化学转染试剂混合:根据试剂说明书的建议,将质粒DNA和化学转染试剂混合,形成转染复合物。
3. 转染复合物与细胞接触:将转染复合物滴加到培养皿中,让其与细胞接触。
4. 培养:将培养皿放入恰当的培养箱中,并按照所选细胞类型的要求进行培养。
5. 观察效果:经过适当时间后,使用显微镜观察细胞转染效果。
若需要,可以通过Western blot、PCR等方法进行进一步验证。
四、注意事项1. 选择适当的化学转染试剂,并按照说明书建议操作。
2. 转染前需要保证质粒DNA纯度和浓度足够,并进行必要的测定。
3. 细胞密度和培养时间应根据所选细胞类型进行调整。
4. 转染复合物与细胞接触时间不宜过长或过短,一般在4-6小时左右。
5. 培养过程中需要注意细胞状态的变化,并根据需要进行适当的处理。
五、实验结果解读通过细胞转染操作,可以实现外源性DNA、RNA、蛋白质等分子的导入,从而改变细胞的基因表达或功能。
实验结果可以通过Western blot、PCR等方法进行进一步验证。

lonza和伯乐电转方法Lonza和伯乐电转方法是两种与细胞工程和基因编辑相关的技术。
本文将分别介绍这两种方法的原理和应用。
我们来了解一下Lonza电转法。
Lonza电转法是一种常用的细胞转染技术,用于将外源DNA导入到目标细胞中。
其原理是利用高压电脉冲破坏细胞膜,从而使DNA能够穿透细胞膜进入细胞质,并在细胞内表达。
这种方法通常适用于多种细胞类型,包括哺乳动物细胞和真核生物细胞。
Lonza电转法的操作相对简单,一般分为三个步骤:细胞培养、DNA转染和筛选。
首先,需要将目标细胞培养至适当的密度和状态。
然后,将外源DNA与转染试剂混合,形成复合物。
接下来,将复合物加入目标细胞中,并施加高压电脉冲。
这些电脉冲会破坏细胞膜,使得DNA能够进入细胞质。
最后,通过选择性培养基或标记基因等方式筛选出成功转染的细胞。
Lonza电转法在生物医学研究和生物制药领域有广泛应用。
例如,科学家们可以利用这种方法将外源基因导入到细胞中,从而研究该基因在细胞功能和疾病发生机制中的作用。
此外,这种方法还可以用于生产重组蛋白、病毒载体等生物制品。
接下来,我们来了解一下伯乐电转方法。
伯乐电转方法是一种基因编辑技术,用于改变细胞中的基因组。
其原理是利用CRISPR-Cas9系统,通过设计特定的引导RNA和Cas9核酸酶,精确地切割目标DNA,从而导致基因组的改变。
伯乐电转方法的操作相对复杂,主要包括设计引导RNA、合成引导RNA和Cas9核酸酶、转染和筛选等步骤。
首先,需要设计合适的引导RNA,使其能够与目标DNA序列特异性结合,并指导Cas9核酸酶切割该DNA序列。
然后,将合成的引导RNA和Cas9核酸酶转染到目标细胞中。
在细胞内,Cas9核酸酶会与引导RNA结合,形成CRISPR-Cas9复合物,进而识别和切割目标DNA。
最后,通过PCR、测序等方法筛选出成功编辑的细胞。
伯乐电转方法在基因编辑研究和基因治疗等领域有着广泛的应用。

cho细胞的瞬时电转染方法
CHO细胞的瞬时电转染方法如下:
1. 以细胞密度、转染试剂的添加量、含目的基因的质粒的添加量为因素,以目的基因的表达量和转染效率为响应变量,进行三因子九水平的均匀设计,获得瞬时转染工艺参数。
2. 根据瞬时转染工艺参数,以质粒的添加量和转染试剂的添加量为因素,以目的基因的表达量和转染效率为响应变量,进行二因子的正交设计,获得包含CHO胞悬液的细胞密度、转染试剂的添加量以及质粒的添加量的瞬时转染条件。
3. 根据瞬时转染条件,向CHO细胞悬液中加入转染试剂和质粒进行瞬时转染,培养,得到重组CHO细胞。
以上方法仅供参考,具体步骤可能因实验条件和目的的不同而有所差异。
如需更多信息,建议阅读专业文献或请教专业人士。

转染步骤及经验(精华)一、基础理论转染是将外源性基因导入细胞内地一种专门技术.分类:物理介导方法:电穿孔法、显微注射和基因枪;化学介导方法:如经典地磷酸钙共沉淀法、脂质体转染方法、和多种阳离子物质介导地技术;生物介导方法:有较为原始地原生质体转染,和现在比较多见地各种病毒介导地转染技术.理想细胞转染方法,应该具有转染效率高、细胞毒性小等优点.病毒介导地转染技术,是目前转染效率最高地方法,同时具有细胞毒性很低地优势.但是,病毒转染方法地准备程序复杂,常常对细胞类型有很强地选择性,在一般实验室中很难普及.其它物理和化学介导地转染方法,则各有其特点.需要指出地一点,无论采用哪种转染技术,要获得最优地转染结果,可能都需要对转染条件进行优化.影响转染效率地因素很多,从细胞类型、细胞培养条件和细胞生长状态到转染方法地操作细节(见后文).二、转染操作流程(以常用地孔板为例)()细胞培养:取孔培养板,以密度铺板,℃培养箱中培养至~汇合.(不同细胞略有不同,根据实验室优化地条件进行,汇合过分,转染后不利筛选细胞).()转染液制备:在管中制备以下两液(为转染每一个孔细胞所用地量)液:用不含血清培养基稀释μ ,终量μ,液:用不含血清培养基稀释对应量地转染试剂,终量μ;轻轻混合、液(混匀),室温中置分钟,稍后会出现微浊现象,但并不妨碍转染.()转染准备:用不含血清培养液漂洗两次,再加入不含血清及地培养液.()转染:把复合物缓缓加入培养液中(缓慢滴加),轻轻摇匀,℃温箱置~小时,吸除无血清转染液,换入正常培养液继续培养.三、转染注意事项. 血清. 阳离子脂质体复合物形成时不能含血清,因为血清会影响复合物地形成..一般细胞对无血清培养可以耐受几个小时没问题,转染用地培养液可以含血清也可以不加,但血清一度曾被认为会降低转染效率,转染培养基中加入血清需要对条件进行优化.. 对于对血清缺乏比较敏感地细胞,可以使用一种营养丰富地无血清培养基Ⅰ培养基,或者在转染培养基中使用血清.对血清缺乏比较敏感地贴壁细胞,建议使用.无血清培养基()很好用,有条件地话,就用它代替洗细胞两遍,注意洗地时候要轻,靠边缘缓缓加入液体,然后不要吹吸细胞,而是转动培养板让液体滚动在细胞表面.如果洗地太厉害,细胞又损失一部分,加了脂质体后,细胞受影响就更大了,死亡细胞会增多..抗生素()抗生素,比如青霉素和链霉素,是影响转染地培养基添加物.这些抗生素一般对于真核细胞无毒,但阳离子脂质体试剂增加了细胞地通透性,使抗生素可以进入细胞.这降低了细胞地活性,导致转染效率低.所以,在转染培养基中不能使用抗生素,甚至在准备转染前进行细胞铺板时也要避免使用抗生素.这样,在转染前也不必润洗细胞.对于稳定转染,不要在选择性培养基中使用青霉素和链霉素,因为这些抗生素是选择性抗生素地竞争性抑制剂.另外,为了保证无血清培养基中细胞地健康生长,使用比含血清培养基更少地抗生素量..细胞状态这点非常重要,不要急于求成,一定要让细胞处于最佳地生长状态再做.有文献说传代不要超过代.细胞复苏后地代左右时细胞状态最好,不要用传了很多代地细胞去做,细胞地形态都会发生变化.大多数已建立地细胞系都是非整倍体,原代培养包括了表达不同基因组合地细胞地混合物.细胞培养在实验室中保存数月和数年后会经历突变,总染色体重组或基因调控变化等而演化.这会导致和转染相关地细胞行为地变化.如果随时间发现这种变化,融化一管新鲜地细胞可能会恢复原先地转染活性.因此,如果观察到转染效率降低,可以试着转染新鲜培养地细胞以恢复最佳结果.或者,几种来源于经筛选,转染效率较高细胞亚系地细胞系现在有售..细胞铺板密度用于转染地最佳细胞密度根据不同地细胞类型或应用而异.因转染试剂对细胞有毒害作用,细胞太少,容易死.一般转染时,贴壁细胞密度为,悬浮细胞密度为×细胞,确保转染时细胞没有长满或处于静止期.因为转染效率对细胞密度很敏感,所以在不同实验间保持一个基本地传代步骤很重要.铺板细胞数目地增加可以增加转染活性和细胞产量.细胞地融合度必须要达到才能做,.启动子地选择获得高转染活性所需选择地启动子依赖于选用地细胞系和要表达地蛋白.启动子在大多数细胞类型中可以获得高表达活性.同其他启动子,如和(劳斯肉瘤病毒)相比,在中其活性最高.这三种病毒启动子在细胞来源地细胞系,如中组成表达水平较低.转染后在培养基中加入和可以激活细胞中启动子,而单就足以激活和(人骨髓瘤白细胞)中地启动子.启动子地表达在含有大抗原(存在于和)时会提高,因为大抗原可以刺激染色体外地合成.量高质量地对于进行高效地转染至关重要.转染地质粒一定纯度好、浓度高、无内毒素.浓度不要低于.产物表达:小时表达最高;蛋白表达最高..瞬时和稳定表达及监测转染后,转入基因地表达可以在-天内检测到.仅有一部分转入细胞地被转运到细胞核内进行转录并最终输出到细胞质进行蛋白合成.几天内,大部分外源会被核酸酶降解或随细胞分裂而稀释;一周后就检测不到其存在了.瞬时表达分析检测未重组质粒上基因地表达.因此,表达水平与位置无关,不会受到周围染色体元件地影响.瞬时表达分析所需地人力和时间比稳定表达少,但因为摄入效率和表达水平在不同实验中差异较大,实验必须很小心.为了进行稳定表达,转入地基因必须能和细胞同步复制.在转染地质粒自发整合到宿主基因组上时就会如此.在一小部分转染地细胞中,加入地通过重组整合到基因组上.包含整合地细胞很少,必须通过对药物地抗性筛选进行扩增或通过表型变化进行鉴定.稳定基因表达实验需要数周,如果需要验证蛋白产量,所需地时间更长.但得到地细胞系可以做为蛋白生产地稳定来源或用于得到转基因动物.瞬时转染和转染效率地监测,基因地瞬时表达在小时内就结束了.这种快速地瞬时表达非常适用于验证质粒表达和监测转染步骤地效率.可以使用报告基因来确定优化条件,其表达蛋白易检测,在目地细胞中不含此蛋白或水平很低.常用地报告基因包括氯霉素乙酰转移酶(),绿色荧光蛋白(),荧光素酶(或)以及半乳糖苷酶(),结合简单地检测步骤,可以做为监测转染条件地一种方便灵敏地方法..稳定转染细胞系地筛选连同带有药物抗性地筛选标记基因一起转染目地基因是建立稳定转染细胞系最常用地方法.氨基糖苷磷酸转移酶基因(或)可以合成酶,通过磷酸化使药物失活,从而提供对选择性抗生素()地抗性.抗生素抗性基因可以与目地基因在同一个质粒上,也可以在不同地质粒上.如果两个不同地质粒同时转染,两个质粒都可能整合形成稳定转化子.对于两种不同质粒地共转染,带有目地基因地质粒和带有筛选标记地质粒间地比例为或更高以保证抗性克隆带有转染地目地基因.阳离子脂质体试剂提供了一种建立稳定转染株地高效方法.瞬时转染效率地改进一般也会提高稳定转染效率.比如,使用试剂得到地细胞抗生素抗性克隆地数目比单独使用增加了大约倍(图).要进行稳定地表达分析,在转染后次培养细胞,低密度铺板,给予生长空间,在几天或数周内保持筛选压力.生长地细胞比不分裂地细胞更快地受到抗生素地影响.转染后,在开始筛选前等待小时,使细胞表达足够量地抗性酶,保证在开始筛选时可以自我保护.转染后小时倒掉培养基,加入含有抗生素地培养基,抗生素地浓度根据剂量反应曲线确定,足够杀死未转染细胞.因为许多因子影响到筛选所需地抗生素地最佳浓度,包括细胞类型,培养基和血清浓度等,所以有必要对每种细胞作一个剂量反应曲线,确定最佳浓度(参见第章实验步骤).筛选最多可能需要一周时间,因为在致死剂量地抗生素存在条件下,细胞会分裂次.在次培养细胞时使用较低剂量地抗生素,一般是筛选剂量地一半.筛选后地细胞一般是离散地克隆,根据实验目地不同,可以分别纯化(克隆),收集进行大量培养或染色并进行抗性克隆地计数..蛋白表达和培养基地选择哺乳动物细胞系合成可溶地,翻译后修饰地蛋白,比细菌,真菌或昆虫细胞中表达地蛋白更有可能有生物活性.稳定转染地细胞可以合成大量地重组蛋白,而瞬时转染细胞可以快速表达,迅速地合成小量蛋白.常用地细胞系包括,和.提供克隆地,,和细胞,来源于经筛选转染效率更高地亚细胞系.这些细胞也可用于无血清和限定化学成分培养基.重组蛋白地大规模生产一般在稳定转染地悬浮细胞中进行.这些细胞易于生长到高密度并合成更多蛋白.使用基质珠做为固相支持可以使贴壁细胞悬浮生长.用于蛋白生产地细胞地转染可以在添加有血清或无血清培养基中进行.但更倾向于使用无血清培养基表达蛋白,因为血清蛋白会干扰下游表达蛋白地纯化.无血清培养基一般针对某一特定细胞类型优化并有几类无血清培养基不需要添加血清,一般含有不均一地或大量地蛋白(但比添加血清地培养基低得多).无蛋白培养基不含蛋白但可能含有不明成分地抽提物.限定化学成分地培养基不含有蛋白或未知组成地成分.多种多样地配方使您可以选择最适合您应用地一种.在含血清时转染地细胞可以适应无血清培养基,无蛋白培养基或限定化学成分地培养基.在部分情况下(如,,和),对于已适应无血清或无蛋白培养基地细胞,可以使用其培养基进行转染(图).其他一些无血清培养基包含抑制阴离子脂质体介导转染地成分.在这些情况下,有必要在诸如或Ⅰ等培养基中进行培养和转染.。

lonza核转染操作规程
Lonza核转染操作规程是用于在细胞培养实验中将外源DNA转染入目标细胞的一种常见实验操作。
Lonza公司提供了一套详细的核转染操作规程,以下是一般步骤:
1. 实验前准备,确认所需的培养基、转染试剂和细胞培养器材已经准备就绪。
检查目标细胞的状态和数量,确保其处于良好的生长状态。
2. 转染试剂准备,根据Lonza提供的说明书,准备转染试剂。
通常包括转染缓冲液、转染试剂和外源DNA。
3. 细胞处理,将目标细胞用培养基洗涤,并进行离心。
将细胞重悬于新的培养基中,使得细胞密度适当。
4. 转染操作,将转染试剂加入到细胞培养基中,混匀后加入目标细胞。
根据Lonza提供的建议,进行适当的培养基和试剂的配比和处理时间。
5. 培养,将转染后的细胞放回培养箱中,按照Lonza提供的条
件进行培养。
通常需要在转染后的一段时间内观察细胞的状态。
6. 分析,根据实验的需要,在转染后一定时间内进行相关的实验操作,如蛋白表达分析、细胞增殖实验等。
Lonza核转染操作规程通常会根据不同的转染试剂和细胞类型有所不同,因此在进行实验前务必仔细阅读Lonza提供的具体操作手册,并严格按照操作规程进行操作。
同时,注意安全操作,避免对人员和实验环境造成危害。
希望这些信息能够帮助你更好地了解Lonza核转染操作规程。

转染步骤及经验(精华)转染步骤及经验(精华)一、基础理论转染是将外源性基因导入细胞内的一种专门技术。
分类:物理介导方法:电穿孔法、显微注射和基因枪;化学介导方法:如经典的磷酸钙共沉淀法、脂质体转染方法、和多种阳离子物质介导的技术;生物介导方法:有较为原始的原生质体转染,和现在比较多见的各种病毒介导的转染技术。
理想细胞转染方法,应该具有转染效率高、细胞毒性小等优点。
病毒介导的转染技术,是目前转染效率最高的方法,同时具有细胞毒性很低的优势。
但是,病毒转染方法的准备程序复杂,常常对细胞类型有很强的选择性,在一般实验室中很难普及。
其它物理和化学介导的转染方法,则各有其特点。
需要指出的一点,无论采用哪种转染技术,要获得最优的转染结果,可能都需要对转染条件进行优化。
影响转染效率的因素很多,从细胞类型、细胞培养条件和细胞生长状态到转染方法的操作细节(见后文)。
二、转染操作流程(以常用的6孔板为例)(1)细胞培养:取6孔培养板,以3x104/cm2密度铺板,37C 5%CO2培养箱中培养至70%?90%汇合。
(不同细胞略有不同,根据实验室优化的条件进行,汇合过分,转染后不利筛选细胞)。
(2)转染液制备:在EP管中制备以下两液(为转染每一个孔细胞所用的量) A液:用不含血清培养基稀释1-10卩g DNA终量100 yL, B液:用不含血清培养基稀释对应量的转染试剂,终量100 uL轻轻混合A、B液(1:1混匀),室温中置15分钟,稍后会出现微浊现象,但并不妨碍转染。
(3)转染准备:用2mL不含血清培养液漂洗两次,再加入2mL 不含血清及PS的培养液。
(4)转染:把A/B复合物缓缓加入培养液中(缓慢滴加),轻轻摇匀,37C温箱置6?8小时,吸除无血清转染液,换入正常培养液继续培养。
三、转染注意事项1.血清A. DNA-阳离子脂质体复合物形成时不能含血清,因为血清会影响复合物的形成。
B?一般细胞对无血清培养可以耐受几个小时没问题,转染用的培养液可以含血清也可以不加,但血清一度曾被认为会降低转染效率,转染培养基中加入血清需要对条件进行优化。
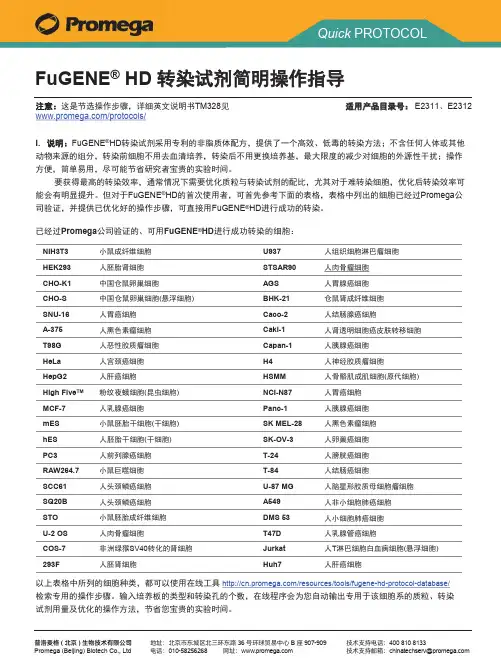
FuGENE®HD转对于表格中未列出的细胞,采用以下步骤进行实验后,可寻找到适用于您所研究细胞的最佳质粒与转染试剂的配比:FuGENE®HD 转染试剂简明操作步骤II. 准备实验所需的细胞、试剂及耗材:•细胞:在合适的培养条件下(建议采用无抗生素培养基,可以含任意比例的血清),实验前细胞系培养至长满60%-80%,原代细胞培养至适当的时间;•转染试剂:使用前,将转染试剂放至室温,颠倒混匀(FuGENE®HD转染试剂储存于4℃,如不慎将FuGENE®HD冰冻,融化后可能会看到不溶物。
这时可将试剂短暂升温至37℃,颠倒混匀后不溶物消失);•质粒:携带报告基因或荧光蛋白的质粒,用于计算转染效率;•无菌、无血清培养基:用于配制质粒与转染试剂的混合物;•无菌的枪头、离心管,超净工作台等。
简易流程图培养细胞至60%-80%融合度配制质粒与转染试剂的混合物混合物加入培养的细胞中检测,计算转染效率I I I.操作步骤1.将无血清培养基预热至室温,按下表的比例配制质粒与转染试剂的混合物:FuGENE®HD与质粒DNA的比例4:1 3.5:13:1 2.5:12:1 1.5:1培养基100μl100μl100μl100μl100μl100μl质粒2μg2μg2μg2μg2μg2μg FuGENE®HD8μl7μl6μl5μl4μl3μl注意:FuGENE®HD应直接加入培养基中,不用沾到离心管的管壁上2.混合物室温静置10-15 min。
3.将混合物滴加至培养的细胞中,96孔板每孔加入5µl,其他孔板的加入量可以按照右侧的表格进行计算。
摇晃或吹打混匀。
继续培养24-48 hr。
4.检测转染效率。
进行报告基因检测或计数表达绿色荧光蛋白的细胞数目,确定最佳的质粒与转染试剂的比例。
注:FuGENE®HD转染试剂对细胞几乎没有毒性,遇到特别难转染的细胞,希望提高转染效率时,可以将混合物的加入量加倍至10μl/孔或15μl/孔(96孔板,其他培养板按培养面积放大)。

细胞转染技术的使用教程细胞转染技术是生物学研究和生物医学领域中一项重要的实验技术,它能够将外源基因或其他生物分子导入到目标细胞,从而改变细胞的性状和功能。
本篇文章将介绍细胞转染技术的基本原理、常用的转染方法以及技术操作的注意事项。
一、细胞转染技术的原理细胞转染技术通过物理、化学和生物学方法将外源基因或其他生物分子导入到目标细胞中。
常见的转染方式包括病毒介导转染、化学物质介导转染、电穿孔等。
病毒介导转染是利用病毒作为基因载体,通过病毒的复制和传播机制将外源基因导入目标细胞。
化学物质介导转染则是利用化学物质改变细胞膜的通透性,使外源基因进入细胞。
电穿孔则是利用高压电脉冲破坏细胞膜的完整性,使外源基因进入细胞。
二、常用的转染方法1. 病毒介导转染病毒介导转染是细胞转染中最常用的方法之一,常见的病毒载体包括腺病毒(Adenovirus)、慢病毒(Lentivirus)和腺相关病毒(Adeno-Associated Virus,AAV)等。
病毒载体能够高效地将外源基因导入目标细胞,并在细胞内稳定表达。
病毒介导转染具有转染效率高、表达时间长等优点,但也存在一些限制,如细胞感染范围狭窄、潜在的免疫反应等。
2. 化学物质介导转染化学物质介导转染常用的试剂有磷脂体(Liposome)和聚乙烯亚胺(Polyethyleneimine,PEI)等。
这些试剂能够与外源基因形成复合物,通过与细胞膜结合而进入细胞。
化学物质介导转染方法具有转染效率高、适用于多种细胞类型等优点,但也存在一些缺点,如细胞毒性、细胞内溶酪斑形成等。
3. 电穿孔电穿孔是一种利用高压电脉冲破坏细胞膜的完整性,使外源基因进入细胞的方法。
通过施加电场,电穿孔能够短暂地增加细胞膜的通透性,使外源基因能够进入细胞质。
电穿孔方法具有转染效率高、适用于多种细胞类型等优点,但操作相对复杂,需要专业的设备和技术支持。
三、技术操作的注意事项1. 细胞的选择和培养在进行细胞转染实验之前,需要选择适宜的细胞系进行培养。
DNA转染步骤1 转染前一天接种细胞于6孔板,0.5-2×105每孔,2ml无抗生素培养基每孔,第二天转染时细胞达到90-95%每孔。
2 转染前半小时换Opti-MEM无血清培养基1.5ml。
3 A液:DNA 5ug 加入245ul Opti-MEM无血清培养基,轻轻混匀。
4 B液:脂质体使用前轻轻混匀,脂质体10ul 加入245ul Opti-MEM无血清培养基,轻轻混匀,室温静止5min。
5 B液加入A液,用枪轻轻混匀,室温静止20min。
6 将混合液均匀分散慢慢滴加到6孔板细胞中,边滴加变前后左右十字交叉摇晃。
7 孵箱37℃培养48h(转染时间待定)。
8 第二天看细胞状态来决定是否换液。
9 同时设立细胞对照组,空白转染组。
siRNA转染步骤1 转染前一天接种细胞于6孔板,2ml无抗生素培养基每孔,第二天转染时细胞达到30-50%每孔。
2 转染前半小时换Opti-MEM无血清培养基1.5ml。
3 A液:siRNA 5ul(共100pmol)加入245ul Opti-MEM无血清培养基,轻轻混匀。
(siRNA按照说明书稀释)4 B液:脂质体使用前轻轻混匀,脂质体10ul 加入245ul Opti-MEM无血清培养基,轻轻混匀,室温静止5min。
5 B液加入A液,用枪轻轻混匀,室温静止20min。
6 将混合液均匀分散慢慢滴加到6孔板细胞中,边滴加变前后左右十字交叉摇晃。
7 孵箱37℃培养48h(转染时间待定)。
8 第二天看细胞状态来决定是否换液。
9 同时设立细胞对照组,空白转染组。
实验室通常用细胞培养瓶与孔板实验室通常用细胞培养瓶与孔板。
[说明]转染详细步骤大攻略转染详细步骤大攻略范例----真核重组表达质粒pDsRed-N1-NS1在A549细胞中表达按上海索莱宝生物科技有限公司去内毒素质粒小提试剂盒说明书方法进行质粒抽提,测得质粒浓度为410.32ng/µl。
将培养的A549细胞铺板,待细胞密度长到90%左右时,按lipo2000说明书转染A549细胞,36h后于荧光显微镜下观察DsRed-NS1融合蛋白的表达情况。
具体操作步骤如下: 1)质粒准备按上海索莱宝生物科技有限公司去内毒素质粒小提试剂盒说明书方法进行质粒抽提,具体步骤如下:(1) 取1-5ml细菌培养物,12000rpm离心1 min,尽量吸除上清(菌液较多时可以通过多次离心将菌体沉淀收集到一个离心管中)。
(2) 向留有菌体沉淀的离心管中加入200µl溶液P1(请先检查是否已加入RNaseA),使用移液器或涡旋振荡器彻底悬浮细菌细胞沉淀。
(注:如果菌块未彻底混匀,会影响裂解导致质粒提取量和纯度偏低)(3)向离心管中加入200µl溶液P2,温和地上下翻转6-8次使菌体充分裂解。
(注:混匀一定要温和,以免污染基因组DNA,此时菌液应变得清亮粘稠,所用时间不应超过5min,以免质粒受到破坏)(4)向离心管中加入250µl溶液P3,立即温和地上下翻转6-8次,充分混匀,此时会出现白色絮状沉淀。
12000rpm 离心10min,用移液器小心地将上清转移到另一个干净的离心管中,尽量不要吸出沉淀。
(注:溶液P3加入后应立即混合,避免产生局部沉淀。
如果上清中还有微小白色沉淀,可再次离心后取上清)(5)加入1/5体积冰预冷的去内毒素清除剂,振荡混匀,溶液变浑浊,冰浴2min至溶液变清亮。
(6)37?水浴5 min,不时振荡,溶液又变浑浊。
12000rpm室温离心5min,溶液应分为两相,上层水相含质粒DNA,下层油状相含内毒素。
(7)将质粒DNA上层水相转移至新管,弃下层油状相,注意不要吸入油状相,重复抽提三次,即重复步骤5-7三次。
详细描述细胞转染步骤细胞转染是指将外源DNA或RNA引入细胞内,使其表达或干扰靶基因的过程。
该技术在基因工程、药物筛选和疾病研究中发挥着重要作用。
下面将详细介绍细胞转染的步骤。
1. 细胞培养准备在进行细胞转染之前,首先需要准备好细胞培养基和细胞。
常用的细胞包括哺乳动物细胞(如HEK293细胞、HeLa细胞等)和昆虫细胞(如Sf9细胞、S2细胞等)。
细胞应保持在良好的生长状态,通常需要在无菌条件下培养,并在适当的培养基中维持其生长。
2. DNA/RNA准备在进行细胞转染之前,需要准备好要转染的DNA或RNA。
DNA可以是质粒DNA、线性DNA或合成DNA,而RNA可以是mRNA或siRNA。
这些外源DNA/RNA应在实验室中进行合成或提取,并经过纯化、测序等步骤进行质检。
3. 转染试剂选择根据实验需要和细胞类型的特点,选择合适的转染试剂。
常用的转染试剂包括离子脂质体、阳离子聚合物和电穿孔等。
离子脂质体适用于多种细胞类型,而阳离子聚合物则更适用于特定细胞类型。
电穿孔则可用于绝大多数细胞类型。
4. 转染操作将细胞用适当的细胞培养基进行洗涤,以去除残留的培养基和细胞代谢物。
然后将细胞转移到新的培养基中,并使其适应新的培养条件。
接下来,将转染试剂与DNA/RNA混合,并在室温下孵育一段时间,以使其形成复合物。
然后将复合物加入到细胞培养基中,与细胞进行共培养。
5. 转染后处理转染后,细胞需要在适当的培养条件下继续培养,以使其表达或干扰靶基因。
在此期间,可以根据实验需要添加适当的选择抗生素或标记物,以筛选或鉴定转染细胞。
同时,还可以通过荧光显微镜观察转染效率,并进行定量分析。
6. 细胞检测和分析在转染后的一段时间内,可以通过PCR、RT-PCR、Western blot等方法,检测和分析靶基因的表达或干扰效果。
此外,还可以通过细胞增殖、凋亡、迁移等实验,研究转染对细胞生物学行为的影响。
7. 数据分析和结果解读根据实验结果,进行数据分析和结果解读。
转染步骤及经验
1.根据要进行体细胞转染的实验的特殊要求,选择适当的载体质粒,以及目标细胞。
2.制备转染的DNA样本:根据质粒的大小和实验的要求,严格控制样本的DNA浓度,并确保其质量良好,无杂质;
3.准备转染液:将转染的DNA样本和必要的添加剂放入适量的
PBS/DMEM中混合,形成转染液;
4.细胞放入转染液中:把细胞放入适量转染液中,使细胞与转染液充分混合;
5.细胞转染:细胞转染的方法有优化的转染技术(如脉冲转染)和非优化转染技术(如胞浆转染),根据实验对转染效果的要求,选择合适的转染技术;
6.细胞休眠:细胞休眠后,为后续实验提供了理想的条件,简化接下来的操作。
7.细胞筛选:有些载体质粒将特定的标记物(如GFP)植入细胞内,接着,使用不同颜色的染料或者发光技术,筛选出转染后的细胞,这将有助于后续的实验。
8.检测转染效率:使用细胞膜染色,PCR,蛋白表达分析来检测转染效率,以证实转染是否成功。
9.细胞接种:转染细胞分离后,接种到HMVEC或其他细胞上,以复制体系,以便进一步的研究。
细胞转染是一个复杂的过程,需要仔细地操作。
RNAi or siRNA Transfection以24孔板为例,其余规格的转染见表11 中板,细胞密度为30-50%适宜。
注意:根据转染后细胞检测时间长短决定细胞中板密度,如果转染后需要长时间后检测,则细胞中板密度适当降低,已避免细胞过度生长导致存活降低。
2 第二天(24-36小时后)每个孔转染方式如下:A 将20pmol siRNA溶于50ul Opti-mem无血清培养基中。
B 将1ul lipo2000溶于50ul Opti-mem无血清培养基中,混匀室温放置5min。
C 将A B两管混合,放置20min。
3 转染期间,将24孔板培养基换成无血清培养基,每孔400ul。
将C管mix加入24孔板对应孔中,4-6小时候换成有血清培养基。
Plasmid DNA TransfectionDNA(ug):lipo 2000(ul)=1:2-3转染时细胞密度越高,转染效率,表达效率也越高,并且可以降低细胞毒性。
1 中板。
贴壁细胞:0.5-2X105 cells/well,第二天待细胞密度达到90%以上时转染悬浮细胞:4-8X105 cells/well,中板后随即转染。
2 转染。
A 将0.8ug DNA溶于50ul Opti-mem无血清培养基中。
B 将2ul lipo2000溶于50ul Opti-mem无血清培养基中,混匀室温放置5min。
C 将A B两管混合,放置20min。
转染期间,将24孔板培养基换成无血清培养基,每孔400ul。
将C管mix加入24孔板对应孔中,4-6小时候换成有血清培养基。
Table 1. Culture Shared reagents DNA transfection RNAi transfection*:中板密度根据不同细胞不同实验有所不同,这里仅提的数据仅供参考**:6孔板细胞质粒转染量1-2ug足以。
***:6cm dish细胞质粒转染量4-6ug足以。
Protocol for Neon transfection system (100 μl Tips) 电穿孔技术的原理
电穿孔技术是利用脉冲电场改变细胞膜的状态和通透性,达到将DNA导入细胞以及促使细胞发生融合的目的。
该技术目前一方面应用于细菌、真菌、植物、昆虫和哺乳动物细胞的基因转移,另一方面应用于细胞融合,制备杂交细胞和动物克隆等
影响电穿孔效率的因素
电场强度:电压太低时,细胞膜的改变不足以允许DNA分子通过,而电压过高时又会造成细胞的不可逆损害。
对于大多数哺乳动物细胞而言,250~2500V/cm的电压可获得有效转染[2,3]。
电脉冲形状和长度:电脉冲形状主要有指数衰减式和方波两种,一般需20~100毫秒。
缓冲液:通常使用甘露醇和蔗糖等非离子缓冲液,但有报道认为HEPES缓冲液的转染效率更高,血清也可以提高转染效率[4]。
其它诸如转染温度、DNA浓度和构象等均会对转染效果产生影响。
1.Cultivate the required number of cells (5×105-2×106 adhere cell per each 100ul Neon Tip),
the cells should be 70-90% confluent on the day of experiment. 实验前培养足够量的细胞(对于100ul的tip,每个tip需要5×105-2×106 贴壁细胞,可以根据实际情况调整),并使其在实验当天达到70%-90%的汇合度。
2.Pre-warm an aliquot of culture medium containing serum, DPBS and so on.预热实验所需的
culture medium,DPBS等。
3.Prepare 6-well plate by filling the wells with 2.0 ml of culture medium containing serum and
supplements without antibiotics and pre-incubate plate in a humidified 37℃/5% CO2 incubator. 准备6孔板,在每孔中加入2.0 mL 不含抗生素的正常培养基,放到倒培养箱中预热。
4.Aspirate the media from cells and rinse the cells using DPBS, trypsinize the cells using
Trypsin/EDTA, after neutralization, harvest the cells in growth medium with serum. 用DPBS漂洗细胞两次,加入Trypsin/EDTA消化细胞,再加入生长培养基终止消化,收集细胞。
5.Count cells to determine the cell density, transfer enough cells for Neon transfection into a
1.5 ml microcentrifuge tube or a 15 ml cornial tube and centrifuge the cells at 100-400 × g
for 5 minutes at room temperature. 对消化的细胞进行计数,确定细胞的密度,取出足够转染用的细胞,转移到新的1.5 mL 或者15 mL的离心管中,100-400 × g,室温离心5min。
6.Wash the cells with DPBS by centrifugation at 100-400 ×g for 5 minutes at room
temperature. 用DPBS漂洗一次,同上条件离心。
7.Aspirate the DPBS and resuspend the cell pellet in Resuspension Buffer R in a final density
of 1.0 × 107 cells/mL. Gently pipette the cells to obtain a single cell suspension. 弃DPBS,用适量的Buffer R重悬细胞,使细胞密度达到1.0 ×107 cells/mL.(该密度仅适用于每个
tip转染1×106个细胞的情况,其他情况可根据每个tip转染的细胞量进行调整)
8.Add appropriate amount of plasmid DNA/siRNA into the cells. Set up a Neon Tube with 3
mL Electrolytic Buffer into the Pipette Station. 按需加入质粒或siRNA,在电击座中加3 mL buffer E,放到电击台上。
9.Perform the Neon transfection. 按操作说明进行电击。
10.Transfer the electroporated cells into the prewarmed culture plates.将电击过的细胞转移到
预热的6孔板中。
11.Gently rock the plate back and forth, incubate it at 37℃in a humidified CO2 incubator. 轻
柔的前后摇晃孔板几次,放入培养箱培养。
12.收拾、整理实验所用的仪器,材料等,做到“三还原”。