双电层
- 格式:docx
- 大小:39.55 KB
- 文档页数:4


双电层理论表面物理化学所涉及的内容非常宽广,固体在溶液中的荷电性质,实际上影响着固体表面性质和界面区的电荷转移反应及其进行的速度。
由于多种极其重要的表面电化学效应的发现,表面电化学引起了许多种科学家的重视和研究。
第一节 双 电 层2.1.1 双电层的产生在自然界中,固体与液体接触时,固体表面的荷电现象实际上是普遍存在的。
它导致了固—液界面的液体一侧带着相反电荷,这种界面电荷影响界面周围介质中的离子分布,与界面电荷符号相反的介质中的离子被吸向界面(这种离子称为反离子Counter -ions ),而相同符号的离子(称为同离子Co -ions )则被排离界面。
与此同时,离子的热运动又促使它们均匀混合在一起。
因此,在带电界面上形成一个扩散双电层(diffuse double layer )。
所谓扩散,就是界面周围介质中的反离子的过量是以扩散形式分布的,而不是非常整齐地集中排列在带电界面的周围。
例如,人体内与血液接触的动静脉壁和血液中胶粒等界面区都存在双电层结构,致使血液在血管中畅通无阻地流动以输送全身新陈代谢的营养而不产生血栓。
双电层理论研究反离子的扩散分布和带电界面的性质。
固体在溶液中荷电而构成双电层的原因,除了外加电场之外,大致上可归纳为以下几种情况: ① 电离作用固体表面在溶液中产生电离或溶液中的电离成分依靠某种结合力与固体表面结合而使其荷电。
例如,玻璃与水接触时,玻璃中的硅酸盐可电离出钾离子、钠离子或氢离子等,于是使玻璃带负电性而溶液带正电性;蛋白质分子具有的羧基(—COOH)和胺基(—NH 2)官能团,当pH 值降低时(酸性),溶液中电离的H +与胺基以氢键结合,从而使蛋白质带正电,-NH 2+H 2O -NH 3++OH -,而溶液一侧带负电,即在羧酸介质中—COOH 的电离被高氢浓度离子所抑制:—COOH+H 2O -COO -+H 3O+ 当pH 值升高时(碱性),蛋白质的羧基电离而使其带负电。


双电层双电层的形成:当两相接触时,如果电子或离子等荷电粒子在两相中具有不同的电化学位,荷电粒子就会在两相之间发生转移或交换,界面两侧便形成符号相反的两层电荷,人们把界面上的这两个荷电层称为双电层。
如金属、溶液界面(M/L)两侧,若μM+>μM+(L),则荷电粒子发生转移,金属表面荷负点;反之,则金属表面荷正,这种双电层常称为离子双层。
尽管有时上述的离子双层并不存在,但金属与溶液界面间仍然会存在着电位差,无论是金属表面,还是溶液表面,都存在着偶极层。
由于偶极子正负电荷分隔开而形成的双电层,称为偶极双电层。
对任何一种金属而言,由于金属的电子会“溢出”金属表面形成双极子。
所以即使溶液一侧不存在偶极子层,但对金属与溶液的界面来说,这种偶极双层总是存在的。
此外,溶液中某一种离子有可能被吸附于电极与溶液界面上,形成一层电荷。
这层电荷又借助静电作用吸引溶液中同等数量的带相反电荷的离子而形成双电层,可称之为吸附双层。
这里应当注意:界面上第一层电荷的出现,靠的是静电力以外的其他化学与物理作用,而第二层电荷则是由第一层电荷的静电力引起的。
如果界面上有了吸附双层,当然也会产生一定大小的电位差。
金属与溶液界面的电位差系由上述的三种类型电位差的一部分或全部组成,但其中对电极反应速度有重大影响的,则主要是离子双层的电位差。
离子双层的形成有两种可能的情况。
一是在电极与溶液一旦接触后的瞬间自发形成的。
另一种情况,是在外电源作用下强制形成的双电层。
因为有的时候,当金属与溶液接触时,并不能自发地形成双电层。
如将纯汞(Hg)放入Kill溶液的界面上常常不能自发的形成双电层。
但是,如果将Hg电极与外电源负极连接,外电源就向Hg电极供应电子,在其电位达到K+还原电位之前,电极上不会发生电化学反应,因而此时Hg电极上有了多余的电子而带上负电。
这层负电荷吸引溶液中相同数量的正电荷(如K+),形成双电层。
双电层的结构模型:金属电极和溶液之间界面上形成的双电层,从结构上可以有离子双电层、表面偶极双电层和吸附双电层等三种类型。


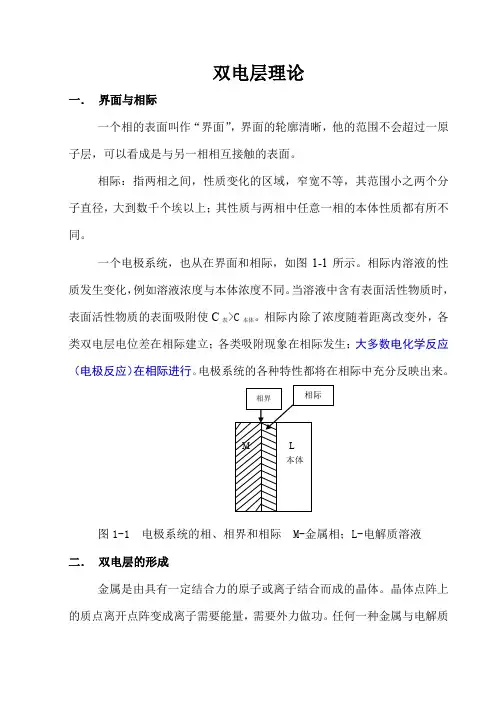
双电层理论一.界面与相际一个相的表面叫作“界面”,界面的轮廓清晰,他的范围不会超过一原子层,可以看成是与另一相相互接触的表面。
相际:指两相之间,性质变化的区域,窄宽不等,其范围小之两个分子直径,大到数千个埃以上;其性质与两相中任意一相的本体性质都有所不同。
一个电极系统,也从在界面和相际,如图1-1所示。
相际内溶液的性质发生变化,例如溶液浓度与本体浓度不同。
当溶液中含有表面活性物质时,表面活性物质的表面吸附使C表>C本体。
相际内除了浓度随着距离改变外,各类双电层电位差在相际建立;各类吸附现象在相际发生;大多数电化学反应(电极反应)在相际进行。
电极系统的各种特性都将在相际中充分反映出来。
图1-1 电极系统的相、相界和相际 M-金属相;L-电解质溶液二.双电层的形成金属是由具有一定结合力的原子或离子结合而成的晶体。
晶体点阵上的质点离开点阵变成离子需要能量,需要外力做功。
任何一种金属与电解质溶液接触时,其界面上的原子(或离子)之间必然发生相互作用,形成双电层。
1.界面电荷层(1)当性质不同的相接触时,在相界面上形成了不同性质的电势差。
(2)出现电势差的原因是带电粒子或偶极子在界面层中的非均匀分布 双电层:由于电极和溶液界面带有的电荷符号相反,故电极/溶液界面上的荷电物质能部分地定向排列在界面两侧。
2.界面电荷层的形成 (1)自发形成的双电层(a )离子双电层 (b )吸附双电层 (c )偶极双电层 (2)强制形成的双电层金属电极与电解质溶液接触,可以自发形成双电层,也可以在外电源作用下强制形成双电层。
以如下电极反应为例:+++++M M2Hg–2e- = Hg22+ ,φ =0.1 VK+ + e- = K , φ= -1.6V 理想极化电极:在一定的电势范围内,可以借助外电源任意改变双电层的带电状况(因而改变界面区的电势差),而不致引起任何电化学反应的电极。
如KCl溶液中的汞电极。
不极化电极:指有电流通过时,电极与溶液界面间电势差不发生任何变化的电极。

双电层理论界面与相际一个相的表面叫作“界面”,界面的轮廓清晰,他的范围不会超过一原子层,可以看成是与另一相相互接触的表面。
相际:指两相之间,性质变化的区域,窄宽不等,其范围小之两个分子直径,大到数千个埃以上;其性质与两相中任意一相的本体性质都有所不同。
一个电极系统,也从在界面和相际,如图1-1所示。
相际内溶液的性质发生变化,例如溶液浓度与本体浓度不同。
当溶液中含有表面活性物质时, 表面活性物质的表面吸附使C a>C本体。
相际内除了浓度随着距离改变外,各类双电层电位差在相际建立;各类吸附现象在相际发生;大多数电化学反应(电极反应)在相际进行。
电极系统的各种特性都将在相际中充分反映出来。
图1-1电极系统的相、相界和相际M-金属相;L-电解质溶液双电层的形成金属是由具有一定结合力的原子或离子结合而成的晶体。
晶体点阵上的质点离开点阵变成离子需要能量,需要外力做功。
任何一种金属与电解质溶液接触时,其界面上的原子(或离子)之间必然发生相互作用,形成双电 层。
1. 界面电荷层(1)当性质不同的相接触时,在相界面上形成了不同性质的电势差。
(2)出现电势差的原因是带电粒子或偶极子在界面层中的非均匀分布双电层:由于电极和溶液界面带有的电荷符号相反,故电极上的荷电物质能部分地定向排列在界面两侧。
2. 界面电荷层的形成(1)自发形成的双电层 (a )离子双电层(2)强制形成的双电层金属电极与电解质溶液接触,可以自发形成双电层,也可以在外电源作 用下强制形成双电层。
以如下电极反应为例:/溶液界面MSM F F干㊀+ (b )吸附双电层 (C )偶极双电层的带电状况(因而改变界面区的电势差),而不致引起任何电化学反应的电 极。
如KCI 溶液中的汞电极。
不极化电极:指有电流通过时,电极与溶液界面间电势差不发生任何变 化的电极。
双电层的建立,引起电位差的变化,这种电位差变化对金属离子继续进 入溶液有阻滞作用,相反有利于返回金属表面。


双电层概念
双电层概念,是电化学中一个非常重要的概念,它是指在电解质溶液中,电极表面与周围溶液之间存在着一层由溶剂分子形成的电性分界层,其厚度为分子级别的层,这个分界层被称为“双电层”。
下面,我们将分步骤来阐述这个概念。
1. 双电层形成的原因
在电解质溶液中,电极表面和周围的溶液之间存在明显的电荷分离现象,这是因为溶剂分子在电极表面的作用下,其电荷状态发生改变,其中,通过电极表面的电子、离子等,与溶液中的带电离子相互作用,在表面附近形成了正负电荷的分离,从而形成了电性分界层,即双电层,这一层在电极表面附近,具有明显的电荷分离现象。
2. 双电层的性质
双电层是由溶剂分子组成的,它具有着一些独特的性质,其中最为特殊的是双电层的厚度非常小,只有分子级别,一般在1nm左右,同时,双电层的电荷密度也非常高,通常在1012~1014 cm-2之间,这些性质决定了双电层与周围环境之间的交互作用非常强烈。
3. 双电层的作用
双电层在电化学中具有着非常重要的作用,其中最为重要的是作为电极与溶液之间的电性分界层,屏蔽了电极表面的电荷,从而保证了电极反应的正常进行。
同时,双电层还能够吸附带电离子和分子,这种吸附作用可以调控电极反应的速率和方向,从而对反应的过程和结果产生深刻的影响。
综上所述,双电层概念是电化学中一个非常重要的概念,它通过精细的电荷分离作用,在电极反应中发挥着至关重要的作用。
双电层的研究不仅能够加深电化学理论的认识,还能够为电化学技术的发展提供新的思路和方法。

电化学界面的基本结构特征双电层
双电层结构主要包括两个重要的部分:电荷层和扩散层。
1.电荷层:
电荷层是电解质离子靠近电极表面的区域,其中的离子成为吸附态离子,形成一个电荷云。
在该区域中,正负电荷的离子分别以吸附在电极表
面并与溶液中的反离子进行排斥。
这些正负离子构成了固、液相之间的分
界面,形成了一个电位差,称为电位的ζ-电位。
电荷层的厚度取决于电解质的浓度和电极的电位。
当电解质浓度低时,电荷层较薄;当电解质浓度高时,电荷层较厚。
同样,当电极电位较高时,电荷层也较厚,而当电极电位较低时,电荷层较薄。
2.扩散层:
扩散层是指离开电极表面的电解质离子的区域。
由于离子在溶液中可
以自由扩散,扩散层中的离子可以自由移动,达到电解质浓度的均匀分布。
在扩散层内,离子的浓度逐渐恢复到远离电极表面时的均匀浓度。
扩散层的厚度取决于电解质的浓度和溶液的流速。
当电解质浓度低或
者溶液的流速高时,扩散层较薄;当电解质浓度高或者溶液的流速低时,
扩散层较厚。
总的来说,电化学界面的基本结构特征是双电层,包括电荷层和扩散层。
电荷层是电解质离子靠近电极表面的区域,其中的离子形成了电位差。
扩散层是离开电极表面的电解质离子的区域,其中离子的浓度会逐渐恢复
到均匀分布。
这些特征对于电化学反应的进行和理解起着重要的作用。
双电层及其结构范文双电层是指电解质溶液与电极表面相接触时,在界面处形成的两层电荷,分别位于溶液和电极之间。
它是电解质溶液与电极之间的过渡区域,是电化学反应的基本界面。
双电层的结构非常复杂,目前尚无一个完全准确的模型来描述其精细的结构,然而,研究表明双电层主要由两个部分构成,即1.电极表面附近的紧密吸附层(Helmholtz层)和2.扩散层。
1. 紧密吸附层(Helmholtz层): 紧密吸附层位于电极表面非常靠近的位置,主要由电极表面附近的离子组成。
它是由于电极表面的电位差吸引了离子,并使离子在电极表面附近紧密吸附而形成。
紧密吸附层中离子的浓度非常高,形成一个电化学的二维层。
Helmholtz层的厚度约为单层离子的直径,通常为纳米级别。
在这一层中,离子以带电的形式吸附在电极表面,形成一个倾斜电场,将离径向分离。
Helmholtz层的电势差称为电解层电势差,它是由于电极表面的电位差引起的。
2. 扩散层: 扩散层位于Helmholtz层外部,主要由水分子和离子组成。
扩散层中的离子浓度逐渐降低,最终达到与体相中离子浓度相同的水平。
扩散层中水分子的运动会受到离子电场的影响,使得水分子的平均速度增加,形成所谓的电场扩散。
在扩散层中,离子的浓度梯度会引起离子的迁移,从而产生扩散电流。
除了紧密吸附层和扩散层,双电层还包括溶液中的离子云层和溶剂分子。
离子云层是离子周围的低浓度区域,通常被认为是由具有相同电荷的反离子组成的。
溶剂分子在双电层中起到辅助离子迁移的作用,与离子之间相互作用。
总结起来,双电层的结构可以简单地描述为电极表面附近的Helmholtz层和外部的扩散层。
Helmholtz层是紧密吸附层,其中的离子以带电形式吸附在电极表面,形成电解层电势差。
扩散层由溶液中的水分子和离子组成,离子在其中通过电场扩散迁移。
双电层的结构是电化学反应的基础,对理解电化学界面和电化学反应机理都具有重要意义。
双电层:(1)决定电位离子层:是固定在胶核表面,并决定其电荷和电位一层离子。
它是由胶体表面的分子解离为离子,或从溶液中吸附某一种离子而构成。
(2)补偿离子层:由于胶体表面决定电位离子层带电,产生电场和静电引力,吸附土壤溶液中带相反电荷的离子,形成补偿离子层。
电荷的来源:1、同晶异质代换作用在自然界中,组成铝硅酸盐矿物晶层的硅四面体和铝八面体中并不完全是硅和铝离子,可以被其他想近或稍大的离子代换。
例如四面体中的硅可被铝代换,八面体中的铝可被铁镁代换。
这种离子代换作用只改变了矿物的化学组成,而矿物的晶体构造形式不变,叫做~。
如硅氧片中的Si4+被Al3+所取代,水铝片中的Al3+被Mg2+、Fe2+所取代,而使晶层产生剩余负电荷。
永久电荷(内电荷):粘粒矿物晶层内的同晶置换所产生的电荷。
电荷数量取决于同晶替代的多少。
特点:不受pH的影响。
2:1型矿物带负电的主要原因。
永久负电荷数量的多少依下规律:蒙脱石、蛭石>水云母类>高岭石2 晶格破碎边缘的断键在矿物风化破碎的过程中,晶体晶格边缘的离子有一部分电荷未得到中和,而产生剩余价键,使晶层带电3 胶核表面分子(或原子团)的解离(1)黏土矿物晶面上-OH的解离(2)腐殖质上某些官能团的解离(COOH)(3)含水铁、铝氧化物的解离(Al2O3.3H2O)(4)含水氧化硅的解离带电:净电荷可变电荷:随ph改变而产生的电荷1:1型矿物特点:晶层与晶层间距离稳定,连接紧密,内部空隙小,电荷量少,单位个体小,分散度低。
多出现于南方酸性土壤,如高岭石类高岭石组黏粒矿物(1:1型矿物)1:1型单位晶层: 由一个硅片和一个铝片构成。
硅片顶端的活性氧与铝片底层的活性氧通过共用的方式形成单位晶层。
这样1:1型层状铝硅酸盐的单位晶层有两个不同的层面,一个是由具有六角形空穴的氧原子层面,一个是由氢氧构成的层面。
包括高岭石、埃洛石、珍珠陶土等特点:(1) 1:1型单位晶胞(层)化学式:Al4Si4O10(OH)8 (2) 膨胀性小晶层间距约0.72nm,硅片和铝片之间存在氢键(3) 电荷数量少同晶替代极少(4)颗粒较大(有效直径0.2~2μm)可塑性、粘结性、吸湿性、粘着性弱2:1型粘土矿物特点:胀缩性大,吸湿性强,易在两边硅氧片中以Al3+代Si4+,有时可在铝氧片中以Mg2+代Al3+→带负电→吸附阳离子。
如蒙脱石,这类矿物多出现于北方土壤。
蒙脱石组黏粒矿物(2:1型矿物)2:1型单位晶层由两个硅片夹一个铝片构成。
两个硅片顶端的氧都向着铝片,铝片上下两层氧分别与硅片通过共用顶端氧的方式形成单位晶层。
这样2:1型层状硅酸盐的单位晶层的两个层面都是氧原子面。
包括蒙脱石、绿脱石、蛭石等特点:(1) 2:1型单位晶胞的理论化学式:Al4Si8O20(OH)4·nH2O (2) 膨胀性大晶层以分子引力联结,晶层间距:蒙脱石0.96~2.14nm 蛭石0.96~1.45nm (3) 电荷数量大同晶替代现象普遍(4) 颗粒较细,呈片状可塑性、粘结性、吸湿性、粘着性显著,对耕作不利蒙脱石在我国北方土壤分布较广,蛭石分布在风化不太强而排水良好的土壤中水化云母组黏粒矿物(2:1型矿物)水化云母(伊利石)组(又称2 :1型非膨胀性矿物)特点:(1) 2 :1型单位晶胞化学式:K2(Al·Fe·Mg)4(Si·Al)8O20(OH)4·nH2O (2) 非膨胀性晶层之间吸附的K+的强吸附力,层间距1.0nm (3) 电荷数量大同晶替代现象普遍,主要发生在硅片,电荷量较大,但部分被层间K+中和,有效电荷量少于蒙脱石(4) 可塑性等性质土壤阳离子交换作用:土壤溶液在一定的ph值是,土壤能吸附的交换性阳离子的总量盐基饱和度:土壤胶体上交换性盐基离子占交换性阳离子总量的百分率盐基离子(Ca2+、Mg2+、K+、Na+、NH4+等)致酸离子H+与Al3+土壤的酸度指土壤酸性的程度,以pH表示。
它是土壤溶液中H+浓度的表现,H+浓度愈大,土壤酸性愈强。
1.活性酸度活性酸度是指土壤溶液中游离氢离子所表现的酸度,通常用pH值来表示2、潜性酸指土壤胶体上吸附的H+或Al3+、Al(OH)2+、Al(OH)2+、Al(OH)2.50.5+所造成的酸性。
因为它们进入溶液后才会显示出酸性,所以称之为潜性酸,常用1000克烘干土中氢离子的厘摩尔数表示。
土壤总碱度:土壤中碳酸根和重碳酸根的总量。
碱化度=(交换性钠/阳离子交换量)x100% 是指交换性钠离子占阳离子交换量的百分数土壤缓冲性酸性或碱性物质加入土壤,土壤具有缓和酸碱反应变化的性能,就是土壤缓冲性,即酸碱缓冲性。
土壤缓冲性产生的原因(1)土壤中有许多弱酸——碳酸、硅酸、磷酸、腐殖酸等,当这些弱酸与其盐类共存,就成为对酸、碱物质具有缓冲作用的体系如HAc+NaAc体系当加入HCl:NaAc+HCl = HAc+NaCl 当加入NaOH: HAc+NaOH = NaAc+H2O2)土壤具有阳离子交换作用主要原因当土壤溶液中H+增加时,胶体表面的交换性盐基离子与溶液中的H+交换,使土壤溶液中H+的浓度基本无变化或变化很小。
当土壤溶液中加入MOH时,解离产生M+或OH-,由于M+与胶体上交换性H+交换,H+转入溶液中,同OH-生成H2O,pH变化极小。
(3)土壤中两性物质的存在(4)酸性土壤中铝离子的缓冲作用在极强酸性土壤中(pH<4),铝以正三价离子状态存在,每个Al3+周围有6个水分子围绕,当加入碱类时,6个水分子中即有一二个解离出H+来中和OH-。
这时带有OH-的铝离子很不稳定,与另一个相同的铝离子结合,在结合中,两个OH-被两个铝离子所共用,并且代替了两个水分子的地位,结果这两个铝离子失去两个正电荷酸性土的调节改良酸性土壤通常施用石灰、石灰石粉和碱性、生理碱性肥料。
碱性土的调节改良碱性土可施用石膏、明矾、硫酸亚铁和硫磺等植物生长发育所需的必需营养元素碳(C)、氢(H)、氧(O)、氮(N)、磷(P)、钾(K)、钙(Ca)、镁(Mg)、硫(S)、铁(Fe)、硼(B)、锰(Mn)、铜(Cu)、锌(Zn)、钼(Mo)和氯(C1)氮素的来源和分布土壤表层的含氮量通常为0.02-0.5%。
地球总氮量的98%存在于地球深处的岩浆岩中,远离我们所生活的土壤-植物-大气-水分环境土壤中的大部分氮素来源于生物固氮、动植物残体的归还、雷电降雨带来的NH4+-N和NO3--N、施入土壤中的化学氮肥和有机肥料硝化作用是指土壤中的铵或氨在微生物的作用下氧化为硝酸盐的过程反硝化作用是指硝酸盐或亚硝酸盐还原为气态氮(分子态氨和氮氧化物)的过程。
铵态氮的固定土壤液相中的铵被土壤颗粒表面所吸附的过程氨的挥发在中性或碱性条件下,土壤中的NH4+转化为NH3而挥发的过程矿化作用(从复杂到简单)土壤有机质在土壤微生物及其酶的作用下,分解成二氧化碳和水,并释放出其中的矿质养分的过程。
腐殖化过程:(从简单到复杂) 土壤有机质在微生物作用下,把有机质分解产生的简单有机化合物及中间产物转化成更复杂的、稳定的、特殊的高分子有机化合物。
植物缺磷抑制体内细胞分裂,使生长缓慢,缺磷体内植物蛋白质合成减慢,有氨基酸积累,同时营养器官内含有大量糖类,有利于叶内合成花青素,使叶子呈紫色、深绿色,根系发育不良,植株表现矮化现象,结实率下。
植株的缺磷首先从老的器官开始。
缺钾症状:从下部老叶开始植株缺钙:生长点坏死中国土壤分类系统分类原则(土纲、亚纲、土类、亚类、土属、土种、亚种)发生学原则:成土因素、成土过程、土壤属性作为基本依据,依土壤属性为基础统一性原则:耕种土壤与自然土壤的统一,具体分析自然因素、人为因素(联系、演变规律)铁铝土纲砖红壤、赤红壤、红壤、黄壤淋溶土纲黄棕壤、黄褐土、棕壤、暗棕壤、白浆土、棕色针叶林土、漂灰土、灰化土半淋溶土纲燥红土、褐土、灰褐土、黑土、灰色森林土干旱土纲灰钙土、棕钙土初育土纲紫色土、龟裂土、新积土、火山灰土、石质土、粗骨土水成土纲沼泽土、泥炭土盐碱土纲滨海盐土、漠境盐土、碱土、草甸盐土、寒原盐土人为土纲水稻土、灌淤土、灌漠土高山土纲(亚)高山草甸土、(亚)高山草原土、山地灌丛草原土、(亚)高山漠土、高山寒漠土中国土壤系统分类分类原则1中国土壤系统分类为共六级,即土纲、亚纲、土类、亚类、土族和土系。
2前四级为高级分类级别,后二级为基层分类级别。
3土纲为最高土壤分类级别,根据主要成土过程产生的或影响主要成土过程的诊断层或/和诊断特性划分。
4亚纲乃土纲的辅助级别,主要以影响现代成土过程的控制因素所反映的诊断特性(如水分状况、温度状况和岩性特性)为划分依据。
5土类则是亚纲的续分,根据反映主要成土过程强度或次要成土过程或次要控制因素的诊断层或/和诊断特性划分。
亚类分类主要依据是否偏离中心概念,是否有附加过程特性和是否有母质残留的特性等土壤分布土壤地带性:经纬度地带性(水平)垂直地带性(海拔)区域地带性(地域性)(地区因素)水平地带性土壤在水平方向上随生物气候带而演替的规律性湿润海洋性带谱∶以秦岭、淮河一线为温带和亚热带的分界线,在此线以南为亚热带和热带地区。
由北向南:棕色森林土--暗棕壤--棕壤--黄棕壤--黄壤--红壤--赤红壤---砖红壤干旱内陆带带谱∶从东到西黑钙土—栗钙土—棕钙土—灰钙土—漠土过渡性土壤带谱∶自黄土高原向东北到大兴安岭西麓褐土—黑垆土—灰褐土—灰黑土—黑土垂直地带性随着海拔高度的变化土壤类型呈现规律性更替的现象东北林区主要土壤类型淋溶土纲:棕色针叶林土、暗棕壤、白浆土半淋溶土纲:黑土半水成土纲:草甸土水成土纲:沼泽土。