notch信号通路幻灯片
- 格式:ppt
- 大小:1.65 MB
- 文档页数:19

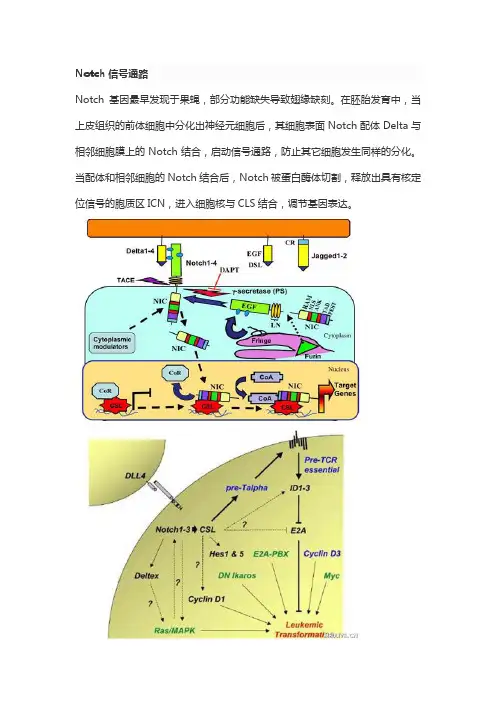
Notch基因最早发现于果蝇,部分功能缺失导致翅缘缺刻(notches,图8-36)。
在胚胎发育中,当上皮组织的前体细胞中分化出神经元细胞后,其细胞表面Notch配体Delta与相邻细胞膜上的Notch结合,启动信号途径,防止其它细胞发生同样的分化,这种现象叫作侧向抑制(lateral inhibition)。
Notch突变的半合子[9]或纯合子在胚胎期死亡,其胚胎中神经组织取代了上皮组织从而使神经组织异常丰富。
Notch信号途径由Notch、Notch配体(DSL蛋白)和CSL(一类DNA结合蛋白)等组成。
Notch及其配体均为单次跨膜蛋白,当配体(如Delta)和相邻细胞的Notch结合后,Notch 被蛋白酶体切割,释放出具有核定位信号的胞质区ICN(intracellular domain of Notch),进入细胞核与CLS结合,调节基因表达。
可概括为(图8-37):Delta→Notch→酶切→ICN→进入细胞核→CLS-ICN复合体→基因转录。
图8-36 Notch缺陷引起果蝇翅缘缺刻Notch:为分子量约300KD的蛋白质,果蝇只有1个Notch基因,人类4个(Notch1-4)。
Notch的胞外区是结合配体的区域,具有不同数量的EGF样重复序列(EGF-R)和3个LNR (Lin/Notch repeats)。
胞内区由RAM(RBP-J kappa associated molecular)结构域、6个锚蛋白(cdc10/ankyrin,ANK)重复序列、2个核定位信号[10](NLS)和PEST结构域。
RAM 结构域是与CSL结合的区域,PEST结构域与Notch的降解有关。
Notch蛋白要经过三次切割,第一次在高尔基体内被furin切割为2个片断,转运到细胞膜形成异二聚体。
当配体结合到胞外区,Notch蛋白又发生两次断裂,先是被肿瘤坏死因子-α-转化酶(TNF-α-converting enzyme,TACE)切割,然后被γ-促分泌酶(γ-secretase)切割,后者需要早老蛋白(presenilin,PS)参与。





Notch信号通路简介命名由来:功能下调会导致果蝇翅膀缺刻。
【1】主要功能参与发育过程中的细胞分化。
参与决定细胞命运。
1.影响果蝇与脊椎动物的神经分化。
在果蝇Notum中,Notch首先确定有分化成神经潜能的细胞的数量(lateral inhibition),再决定这些细胞的后代中哪些分化成神经,哪些分化成神经胶质(lineage decisions)。
2.果蝇翅膀中Notch信号通路决定D-V界限,它的缺失可能引起翅的缺刻。
2龄幼虫开始形成背腹间隔,选择基因Ap(apterous)在翅膀的背区表达,诱导Fringe 和Serrate 在背隔间区表达,而Delta则在背腹区均有表达。
在背间隔区,Fringe抑制serrate的功能,而促进Dl的功能。
所以,serrate在靠近DV界限的腹间隔区激活Notch(没有fringe),而Dl在靠近DV界限的背间隔区激活Notch(fringe激活其活性)。
Notch信号可能与癌症相关。
50% 的 T-cell acute lymphoblastic leukaemias中都可以检测到Notch 1的突变。
Notch还参与调控血管的生成。
Notch信号可能与免疫相关。
它可以促进Tαβ细胞的形成,与Gata 3基因协同调控 CD4+细胞向Th 1/Th 2 类型的分化[7],并且可增加外周免疫器官边缘区B 细胞的数量。
¤- Notch信号通路只能影响相邻的细胞。
没有二级信使,信号传递速度快。
¤-相邻细胞可以通过Notch受体与配体的结合传递Notch信号,从而扩大并固化细胞间的分子差异,最终决定细胞命运,影响器官形成和形态发生。
¤-Notch信号在细胞中常被反复激活,决定不同的细胞命运。
(比如神经细胞的分化、比如翅形态建成)【2】信号通路的成员Serrate (Jagged1、Jagged2 in mammals) ★功能:Notch配体,激活受体细胞Notch信号通路。



Notch信号通路Notch基因最早发现于果蝇,部分功能缺失导致翅缘缺刻。
在胚胎发育中,当上皮组织的前体细胞中分化出神经元细胞后,其细胞表面Notch配体Delta与相邻细胞膜上的Notch结合,启动信号通路,防止其它细胞发生同样的分化。
当配体和相邻细胞的Notch结合后,Notch被蛋白酶体切割,释放出具有核定位信号的胞质区ICN,进入细胞核与CLS结合,调节基因表达。
composed of a short extracellular region, a single transmembrane-pass, and aNotch-mediated juxtacrine signal between adjacent cells.Notch signaling stepsNotch信号通路抑制剂100年以前,Thomas Hunt Morgan发现一种基因突变会导致黑腹果蝇锯齿缘翅膀(notched wing),于是将这个基因命名为Notch,Notch信号通路的研究也就是从这里起步。
现在Notch信号异常已经与多种疾病联系起来,包括肿瘤、CADASIL综合征、Alagille综合征、脊椎肋骨发育不全,Notch信号通路抑制剂已经进入临床研究阶段。
Notch是细胞之间的通讯系统,信息在信号发出细胞与信号接收细胞之间传递,维持组织的发育和稳态。
Notch受体有Notch1、Notch2、Notch3、Notch4四种,而Notch配体有DLL1、DLL3、DLL4、JAG1、JAG2五种,当配体与受体相互作用,Notch受体经过三次剪切,胞内区(NICD)释放出来进入细胞核,激活相关基因转录。
目前的研究发现,造血系统、脉管系统、心脏发育、皮肤分化、脊柱、内耳、肝脏、肿瘤等与Notch信号有关,肿瘤是目前的研究热点,乳腺癌、T细胞白血病等存在Notch1激活/强化突变。
Notch信号调节动脉、静脉发育的平衡,抑制Notch信号会打破脉管系统平衡,肿瘤得不到有效供血,这是一种可能的抗肿瘤机制。

•·NGF信号通路(2004-8-16)•·TGF beta 信号转导(2004-8-16)•·细胞凋亡信号(2004-8-16)•·线粒体输入信号(2004-8-16)•·ROS信号(2004-8-16)•·Toll-Like 受体家族(2004-8-16)•·Toll-Like 受体(2004-8-16)•·actin肌丝(2004-8-16)•·Wnt/LRP6 信号(2004-8-16)•·WNT信号转导(2004-8-16)•·West Nile 西尼罗河病毒(2004-8-16)•·Vitamin C 维生素C在大脑中的作用(2004-8-16)•·视觉信号转导(2004-8-16)•·VEGF,低氧(2004-8-16)•·TSP-1诱导细胞凋亡(2004-8-16)•·Trka信号转导(2004-8-16)•·dbpb调节mRNA (2004-8-16)•·CARM1甲基化(2004-8-16)•·CREB转录因子(2004-8-16)•·TPO信号通路(2004-8-16)•·Toll-Like 受体(2004-8-16)•·TNFR2 信号通路(2004-8-16)•·TNFR1信号通路(2004-8-16)•·TNF/Stress相关信号(2004-8-16)•·IGF-1受体(2004-8-16)•·共刺激信号(2004-8-16)•·Th1/Th2 细胞分化(2004-8-16)•·TGF beta 信号转导(2004-8-16)•·端粒、端粒酶与衰老(2004-8-16)•·TACI和BCMA调节B细胞免疫(2004-8-16)•·T辅助细胞的表面受体(2004-8-16)•·T细胞受体信号通路(2004-8-16)•·T细胞受体和CD3复合物(2004-8-16)•·Cardiolipin的合成(2004-8-16)•·Synaptic突触连接中的蛋白(2004-8-16)•·HSP在应激中的调节的作用(2004-8-16)•·Stat3 信号通路(2004-8-16)•·SREBP控制脂质合成(2004-8-16)•·酪氨酸激酶的调节(2004-8-16)•·Sonic Hedgehog (SHH)受体ptc1调节细胞周期(2004-8-16)•·Sonic Hedgehog (Shh) 信号(2004-8-16)•·SODD/TNFR1信号(2004-8-16)•·AKT/mTOR在骨骼肌肥大中的作用(2004-8-16)•·G蛋白信号转导(2004-8-16)•·肝细胞生长因子受体信号(2004-8-16)•·IL1受体信号转导(2004-8-16)•·acetyl从线粒体到胞浆过程(2004-8-16)•·趋化因子chemokine在T细胞极化中的选择性表(2004-8-16)•·SARS冠状病毒蛋白酶(2004-8-16)•·Parkin在泛素-蛋白酶体中的作用(2004-8-16)•·nicotinic acetylcholine受体在凋亡中的作用(2004-8-16)•·线粒体在细胞凋亡中的作用(2004-8-16)•·MEF2D在T细胞凋亡中的作用(2004-8-16)•·Erk5和神经元生存(2004-8-16)•·ERBB2信号转导(2004-8-16)•·GPCRs调节EGF受体(2004-8-16)•·BRCA1调节肿瘤敏感性(2004-8-16)•·Rho细胞运动的信号(2004-8-16)•·Leptin能逆转胰岛素抵抗(2004-8-16)•·转录因子DREAM调节疼敏感(2004-8-16)•·PML调节转录(2004-8-16)•·p27调节细胞周期(2004-8-16)•·MAPK信号调节(2004-8-16)•·细胞因子调节造血细胞分化(2004-8-16)•·eIF4e和p70 S6激酶调节(2004-8-16)•·eIF2调节(2004-8-16)•·谷氨酸受体调节ck1/cdk5 (2004-8-16)•·plk3在细胞周期中的作用(2004-8-1)•·BAD磷酸化调节(2004-8-1)•·Reelin信号通路(2004-8-1)•·RB肿瘤抑制和DNA破坏(2004-8-1)•·NK细胞介导的细胞毒作用(2004-8-1)•·Ras信号通路(2004-8-1)•·Rac 1细胞运动信号(2004-8-1)•·PTEN依赖的细胞生长抑制和细胞凋亡(2004-8-1)•·notch信号通路(2004-8-1)•·蛋白激酶A(PKA)在中心粒中的作用(2004-8-1)•·蛋白酶体Proteasome复合物(2004-8-1)•·Prion朊病毒的信号通路(2004-8-1)•·早老素Presenilin在notch和wnt信号中的作用(2004-8-1)•·mRNA的poly(A)形成(2004-8-1)•·淀粉样蛋白前体信号(2004-8-1)•·PKC抑制myosin磷酸化(2004-8-1)•·磷脂酶C(PLC)信号(2004-8-1)•·巨噬细胞Pertussis toxin不敏感的CCR5信号通(2004-8-1)•·Pelp1调节雌激素受体的活性(2004-8-1)•·PDGF信号通路(2004-8-1)•·p53信号通路(2004-8-1)•·p38MAPK信号通路(2004-8-1)•·Nrf2是氧化应激基本表达的关键基因(2004-8-1)•·OX40信号通路(2004-8-1)•·hTerc转录调节活性图(2004-8-1)•·hTert转录因子的调节作用(2004-8-1)•·AIF在细胞凋亡中的作用(2004-8-1)•·Omega氧化通路(2004-8-1)•·核受体在脂质代谢和毒性中的作用(2004-8-1)•·NK细胞中NO2依赖的IL-12信号通路(2004-8-1)•·TOR信号通路(2004-8-1)•·NO信号通路(2004-8-1)•·NF-kB信号转导通路(2004-8-1)•·NFAT与心肌肥厚的示意图(2004-8-1)•·神经营养素及其表面分子(2004-8-1)•·神经肽VIP和PACAP防止活化T细胞凋亡图(2004-8-1)•·神经生长因子信号图(2004-8-1)•·线虫和哺乳动物的MAPK信号比较(2004-7-17)•·细胞内信号总论(2004-7-17)•·细胞凋亡信号通路(2004-7-17)•·MAPK级联通路(2004-7-17)•·MAPK信号通路图(2004-7-17)•·BCR信号通路(2004-7-17)•·蛋白质乙酰化示意图(2004-7-17)•·wnt信号通路(2004-7-17)•·胰岛素受体信号通路(2004-7-17)•·细胞周期在G2/M期的调控机理图(2004-7-17)•·细胞周期G1/S检查点调控机理图(2004-7-17)•·Jak-STAT关系总表(2004-7-17)•·Jak/STAT 信号(2004-7-17)•·TGFbeta信号(2004-7-17)•·NFkappaB信号(2004-7-17)•·p38 MAPK信号通路(2004-7-17)•·SAPK/JNK 信号级联通路(2004-7-17)•·从G蛋白偶联受体到MAPK (2004-7-17)•·MAPK级联信号图(2004-7-17)•·eIF-4E和p70 S6激酶调控蛋白质翻译(2004-7-17)•·eif2蛋白质翻译(2004-7-17)•·蛋白质翻译示意图(2004-7-17)•·线粒体凋亡通路(2004-7-17)•·死亡受体信号通路(2004-7-17)•·凋亡抑制通路(2004-7-17)•·细胞凋亡综合示意图(2004-7-17)•·Akt/Pkb信号通路(2004-7-17)•·MAPK/ERK信号通路(2004-7-17)•·哺乳动物MAPK信号通路(2004-7-17)•·Pitx2多步调节基因转录(2004-7-17)•·IGF-1R导致BAD磷酸化的多个凋亡路径(2004-7-17)•·多重耐药因子(2004-7-17)•·mTOR信号通路(2004-7-17)•·Msp/Ron受体信号通路(2004-7-17)•·单核细胞和其表面分子(2004-7-17)•·线粒体的肉毒碱转移酶(CPT)系统(2004-7-17)•·METS影响巨噬细胞的分化(2004-7-17)•·Anandamide,内源性大麻醇的代谢(2004-7-17)•·黑色素细胞(Melanocyte)发育和信号(2004-7-17)•·DNA甲基化导致转录抑制的机理图(2004-7-17)•·蛋白质的核输入信号图(2004-7-17)•·PPARa调节过氧化物酶体的增殖(2004-7-17)•·对乙氨基酚(Acetaminophen)的活性和毒性机(2004-7-17)•·mCalpain在细胞运动中的作用(2004-7-17)•·MAPK信号图(2004-7-17)•·MAPK抑制SMRT活化(2004-7-17)•·苹果酸和天门冬酸间的转化(2004-7-17)•·低密度脂蛋白(LDL)在动脉粥样硬化中的作用(2004-7-17)•·LIS1基因在神经细胞的发育和迁移中的作用图(2004-7-17)•·Pyk2与Mapk相连的信号通路(2004-7-17)•·galactose代谢通路(2004-7-17)•·Lectin诱导补体的通路(2004-7-17)•·Lck和Fyn在TCR活化中的作用(2004-7-17)•·乳酸合成图(2004-7-17)•·Keratinocyte分化图(2004-7-17)•·离子通道在心血管内皮细胞中的作用(2004-7-17)•·离子通道和佛波脂(Phorbal Esters)信号(2004-7-17)•·内源性Prothrombin激活通路(2004-7-17)•·Ribosome内化通路(2004-7-17)•·整合素(Integrin)信号通路(2004-7-17)•·胰岛素(Insulin)信号通路(2004-7-17)•·Matrix Metalloproteinases (2004-7-17)•·组氨酸去乙酰化抑制剂抑制Huntington病(2004-7-17)•·Gleevec诱导细胞增殖(2004-7-17)•·Ras和Rho在细胞周期的G1/S转换中的作用(2004-7-17)•·DR3,4,5受体诱导细胞凋亡(2004-7-17)•·AKT调控Gsk3图(2004-7-17)•·IL-7信号转导(2004-7-17)•·IL22可溶性受体信号转导图(2004-7-17)•·IL-2活化T细胞图(2004-7-17)•·IL12和Stat4依赖的TH1细胞发育信号通路(2004-7-17)•·IL-10信号通路(2004-7-17)•·IL 6信号通路(2004-7-17)•·IL 5信号通路(2004-7-17)•·IL 4信号通路(2004-7-17)•·IL 3信号通路(2004-7-17)•·IL 2 信号通路(2004-7-17)•·IL 18信号通路(2004-7-17)•·IL 17信号通路(2004-7-17)•·IGF-1信号通路(2004-7-17)•·IFN gamma信号通路(2004-7-17)•·INF信号通路(2004-7-17)•·低氧诱导因子(HIF)在心血管中的作用(2004-7-17)•·低氧和P53在心血管系统中的作用(2004-7-17)•·人类巨细胞病毒和MAP信号通路(2004-7-17)•·孕酮如何促进卵细胞成熟?(2004-7-17)•·How does salmonella hijack a cell (2004-7-17)•·Hop通路在心脏发育中的作用(2004-7-17)•·HIV-I Nef:负性调节fas和TNF (2004-7-17)•·HIV-1防止宿主细胞耐受的机理(2004-7-17)•·HIV诱导T细胞凋亡图(2004-7-17)•·血红素的伴侣分子(2004-7-17)•·g-Secretase介导ErbB4信号通路(2004-7-17)•·生物激素信号(2004-7-17)•·Granzyme A介导的凋亡信号通路(2004-7-17)•·G蛋白偶联信号需要Tubby支持(2004-7-17)•·糖酵解通路(2004-7-17)•·Ghrelin:食物吸收和能量平衡的调控者(2004-7-17)•·PS1能产生beta淀粉样蛋白导致老年性痴呆(2004-7-17)•·GATA3部分参与TH2细胞因子基因的表达(2004-7-17)•·GABA受体的代谢图(2004-7-17)•·FXR和LXR调节胆固醇代谢(2004-7-17)•·SLRP在骨骼中的作用(2004-7-17)•·自由基诱导细胞凋亡信号(2004-7-17)•·FOSB与药物成瘾(2004-7-17)•·fMLP诱导趋化因子基因表达(2004-7-17)•·Fibrinolysis通路(2004-7-17)•·糖酵解通路(2004-7-17)•·Fc Epsilon Receptor I信号(2004-7-17)•·FAS信号通路(2004-7-17)•·外源性Prothrombin激活通路(2004-7-17)•·真核细胞蛋白质翻译示意图(2004-7-17)•·雌激素反应蛋白EFP控制乳腺癌细胞的细胞周期(2004-7-17)•·EPO介导神经保护作用与NF-kB相关(2004-7-17)•·Erythrocyte分化通路(2004-7-17)•·Erk1/Erk2 Mapk 信号通路(2004-7-17)•·Erk和PI-3K在细胞外间质中的作用(2004-7-17)•·内质网相关的蛋白质降解通路示意图(2004-7-17)•·EPO售转导机制图(2004-7-17)•·血小板凝聚示意图(2004-7-17)•·NDK动力学(2004-7-17)•·线粒体的电子传递链示意图(2004-7-17)•·Eicosanoid代谢(2004-7-17)•·EGF信号通路(2004-7-17)•·calcineurin对Keratinocyte分化的影响(2004-7-17)•·E2F1信号通路(2004-7-17)•·MTA-3在雌激素不敏感性乳腺癌中下调(2004-7-17)•·双链RNA诱导基因表达示意图(2004-7-17)•·Dicer信号通路(RNAi机理)(2004-7-17)•·CDK5在老年性痴呆中的调节作用(2004-7-17)•·树突状细胞调节TH1和TH2发育示意图(2004-7-17)•·RAR和RXR被蛋白酶体降解通路(2004-7-17)•·D4-GDI信号通路示意图(2004-7-17)•·细胞因子和炎症反应示意图(2004-7-9)•·细胞因子网络调控图(2004-7-9)•·CFTR和beta 2肾上腺素受体通路(2004-7-9)•·Cyclin和细胞周期调控图(2004-7-9)•·Ran核质循环转运图(2004-7-9)•·Cyclin E降解通路图(2004-7-9)•·CXCR4信号通路图(2004-7-9)•·CTL介导的免疫反应图(2004-7-9)•·CTCF:第一个多价核因子(2004-7-9)•·皮质激素和心脏保护(2004-7-9)•·骨骼肌的成肌信号图(2004-7-9)•·VitD调控基因表达信号图(2004-7-9)•·补体信号通路(2004-7-9)•·线粒体和过氧化物酶体中β氧化的比较图(2004-7-9)•·经典的补体信号通路图(2004-7-9)•·心律失常的分子机制图(2004-7-9)•·hSWI/SNF ATP依赖的染色体重塑(2004-7-9)•·碳水化合物和cAMP调节ChREBP图(2004-7-9)•·分子伴侣调节干扰素信号图(2004-7-9)•·Ceramide信号图(2004-7-9)•·局部急性感染的细胞与分子信号(2004-7-9)•·细胞与细胞粘附信号(2004-7-9)•·细胞周期G2/M调控点信号调节(2004-7-9)•·细胞周期 G1/S调控点信号图(2004-7-9)•·CDK调节DNA复制(2004-7-9)•·cdc25和chk1在DNA破坏中的作用图(2004-7-9)•·CD40L信号通路图(2004-7-9)•·CCR3信号图(2004-7-9)•·CBL下调EGF受体的信号转导图(2004-7-9)•·一些氨基酸的代谢通路图 3 (2004-7-9)•·一些氨基酸的代谢通路图 2 (2004-7-9)•·一些氨基酸的代谢通路图(2004-7-9)•·Catabolic pathway for asparagine and asp (2004-7-9)•·Caspase 信号级联通路在细胞凋亡中的作用(2004-7-9)•·CARM1和雌激素的信号转导调控(2004-7-9)•·抗氧自由基的心脏保护作用信号转导图(2004-7-9)•·乙肝病毒中的钙信号调控(2004-7-9)•·镉诱导巨噬细胞的DNA合成和增殖(2004-7-9)•·Ca2+/CaM依赖的激活(2004-7-9)•·B细胞活化机理图(2004-6-9)•·BTG家族蛋白和细胞周期的调节(2004-6-9)•·BRCA1作用机理(2004-6-9)•·骨重塑示意图(2004-6-9)•·Botulinum Toxin阻断神经递质释放示意图(2004-6-9)•·缬氨酸的生物合成图(2004-6-9)•·Tryptophan在植物和细菌内的生物合成(2004-6-9)•·苏氨酸和蛋氨酸的体内合成示意图(2004-6-9)•·sphingolipids生物合成(2004-6-9)•·spermidine和spermine生物合成(2004-6-9)•·细菌体内合成脯氨酸的示意图(2004-6-9)•·苯丙氨酸和酪氨酸的生物合成(2004-6-9)•·神经递质的合成示意图(2004-6-9)•·赖氨酸生物合成图(2004-6-9)•·亮氨酸的体内生物合成图(2004-6-9)•·异亮氨酸的生物合成图(2004-6-9)•·甘氨酸和色氨酸的生物合成(2004-6-9)•·Cysteine在哺乳动物中的合成图(2004-6-9)•·Cysteine在细菌和植物内生物合成图(2004-6-9)•·Chorismate在细菌和植物内的生物合成(2004-6-9)•·Arginine在细菌内的生物合成(2004-6-9)•·生物活性肽诱导的通路(2004-6-9)•·脂肪酸的β氧化通路(2004-6-9)•·BCR信号通路示意图(2004-6-9)•·SUMOylation基本机理(2004-6-9)•·PPAR影响基因表达的基本信号机制图(2004-6-9)•·B淋巴细胞表面分子示意图(2004-6-9)•·B细胞生存信号通路(2004-6-5)•·B细胞信号通路的复杂性(2004-6-5)•·GPCR信号的衰减的机理(2004-6-4)•·ATM信号通路(2004-6-4)•·阿斯匹林的抗凝机理(2004-6-4)•·细胞凋亡信号调节DNA片段化(2004-6-4)•·细胞凋亡DNA片段化与组织稳态的机理(2004-6-4)•·反义核酸的作用机理---RNA polymerase III (2004-6-4)•·抗原递呈与处理信号图(2004-6-4)•·Antigen依赖的B细胞激活(2004-6-4)•·Anthrax Toxin Mechanism of Action (2004-6-4)•·血管紧张素转换酶2调节心脏功能(2004-6-4)•·Angiotensin II 介导JNK信号通路的激活(2004-6-4)•·Alternative Complement Pathway (2004-6-4)•·Alpha-synuclein和Parkin在怕金森病中的作用(2004-6-4)•·ALK在心肌细胞中的功能图(2004-6-4)•·AKT信号通路(2004-6-4)•·AKAP95在有丝分裂中的作用图(2004-6-4)•·Ahr信号转导图(2004-6-4)•·Agrin突触后的功能图(2004-6-4)•·ADP-Ribosylation 因子(2004-6-4)•·淋巴细胞粘附分子信号图(2004-6-4)•·Adhesion and Diapedesis of Lymphocytes (2004-6-4)•·Adhesion and Diapedesis of Granulocytes (2004-6-4)•·急性心肌梗死信号转导图(2004-6-4)•·src蛋白质激活图(2004-6-4)•·PKC与G蛋白耦联受体的关系(2004-6-4)•·cAMP依赖的CSK抑制T细胞功能示意图(2004-6-4)•·PKA功能示意图(2004-6-4)•·一氧化氮(NO)在心脏中的功能示意图(2004-6-4)•·RelA 在细胞核内乙酰化和去乙酰化(2004-6-4)actin肌丝Mammalian cell motility requires actin polymerization in the direction of movement to change membrane shape and extend cytoplasm into lamellipodia. The polymerization of actin to drive cell movement also involves branching of actin filaments into a network oriented with the growing ends of the fibers near the cell membrane. Manipulation of this process helps bacteria like Salmonella gain entry into cells they infect. Two of the proteins involved in the formation of Y branches and in cell motility are Arp2 and Arp3, both members of a large multiprotein complex containing several other polypeptides as well. The Arp2/3 complex is localized at the Y branch junction and induces actin polymerization. Activity of this complex is regulated by multiple different cell surface receptor signaling systems, activating WASP, and Arp2/3 in turn to cause changes in cell shape and cell motility. Wasp and its cousin Wave-1 interact with the Arp2/3 complex through the p21 component of the complex. The crystal structure of the Arp2/3 complex has revealed further insights into the nature of how the complex works.Activation by Wave-1, another member of the WASP family, also induces actin alterations in response to Rac1 signals upstream. Wave-1 is held in an inactive complex in the cytosol that is activated to allow Wave-1 to associate with Arp2/3. While WASP is activated by interaction with Cdc42, Wave-1, is activated by interaction with Rac1 and Nck. Wave-1 activation by Rac1 and Nck releases Wave-1 with Hspc300 to activate actin Y branching and polymerization by Arp2/3. Different members of this gene family may produce different actin cytoskeletal architectures. The immunological defects associated with mutation of the WASP gene, theWiskott-Aldrich syndrome for which WASP was named, indicates the importance of this system for normal cellular function.Cory GO, Ridley AJ. Cell motility: braking WAVEs. Nature. 2002 Aug 15;418(6899):732-3. No abstract available.Eden, S., et al. (2002) Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418(6899), 790-3Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp. Blood. 2002 Sep 15;100(6):2113-22.Kreishman-Deitrick M, Rosen MK, Kreishman-Deltrick M. Ignition of a cellular machine. Nat Cell Biol. 2002 Feb;4(2):E31-3. No abstract available.Machesky, L.M., Insall, R.H. (1998) Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol 8(25), 1347-56Robinson, R.C. et al. (2001) Crystal structure of Arp2/3 complex. Science 294(5547), 1679-84Weeds A, Yeoh S. Structure. Action at the Y-branch. Science. 2001 Nov 23;294(5547):1660-1. No abstract available.Wnt/LRP6 信号Wnt glycoproteins play a role in diverse processes during embryonic patterning in metazoa through interaction with frizzled-type seven-transmembrane-domain receptors (Frz) to stabilize b-catenin. LDL-receptor-related protein 6 (LRP6), a Wnt co-receptor, is required for this interaction. Dikkopf (dkk) proteins are both positive and negative modulators of this signalingWNT信号转导West Nile 西尼罗河病毒West Nile virus (WNV) is a member of the Flaviviridae, a plus-stranded virus family that includes St. Louis encephalitis virus, Kunjin virus, yellow fever virus, Dengue virus, and Japanese encephalitis virus. WNV was initially isolated in 1937 in the West Nile region of Uganda and has become prevalent in Africa, Asia, and Europe. WNV has rapidly spread across the United States through its insect host and causes neurological symptoms and encephalitis, which can result in paralysis or death. Since 1999 about 3700 cases of West Nile virus (WNV) infection and 200 deaths have been recorded in United States. The viral capsid protein likely contributes to the WNV-associated deadly inflammation via apoptosis induced through the mitochondrial pathway.WNV particles (50 nm in diameter) consist of a dense core (viral protein C encapsidated virus RNA genome)surrounded by a membrane envelope (viral E and M proteins embedded in a lipid bilayer). The virus binds to a specific cell surface protein (not yet identified), an interaction thought to involve E protein with highly sulfated neperan sulfate (HSHS) residues that are present on the surfaces of many cells and enters the cell by a process similar to that of endocytosis. Once inside the cell, the genome RNA is released into the cytoplasm via endosomal release, a fusion process involving acidic pH induced conformation change in the E protein. The RNA genome serves as mRNA and is translated by ribosomes into ten mature viral proteins are produced via proteolytic cleavage, which include three structural components and seven different nonstructural components of the virus. These proteins assemble and transcribe complimentary minus strand RNAs from the genomic RNA. The complimentary minus strand RNA in turns serves as template for the synthesis of positive-stranded genomic RNAs. Once viral E, preM and C proteins have accumulated to sufficient level, they assemble with the genomic RNA to form progeny virions, which migrate to the cell surface where they are surrounded with lipid envelop and released.Vitamin C 维生素C在大脑中的作用Vitamin C (ascorbic acid) was first identified by virtue of the essential role it plays in collagen modification, preventing the nutritional deficiency scurvy. Vitamin C acts as a cofactor for hydroxylase enzymes thatpost-translationally modify collagen to increase the strength and elasticity of tissues. Vitamin C reduces the metal ion prosthetic groups of many enzymes, maintaining activity of enzymes, also acts as an anti-oxidant. Although the prevention of scurvy through modification of collagen may be the most obvious role for vitamin C, it is not necessarily the only role of vitamin C. Svct1 and Svct2 are ascorbate transporters for vitamin C import into tissues and into cells. Both of these transporters specifically transport reduced L-ascorbic acid against a concentration gradient using the intracellular sodium gradient to drive ascorbate transport. Svct1 is expressed in epithelial cells in the intestine, upregulated in cellular models for intestinal epithelium and appears to be responsible for the import of dietary vitamin C from the intestinal lumen. The vitamin C imported from the intestine is present in plasma at approximately 50 uM, almost exclusively in the reduced form, and is transported to tissues to play a variety of roles. Svct2 imports reduced ascorbate from the plasma into veryactive tissues like the brain. Deletion in mice of the gene for Svct2 revealed that ascorbate is required for normal development of the lungs and brain during pregnancy. A high concentration of vitamin C in neurons of the developing brain may help protect the developing brain from free radical damage. The oxidized form of ascorbate, dehydroascorbic acid, is transported into a variety of cells by the glucose transporter Glut-1. Glut-1, Glut-3 and Glut-4 can transport dehydroascorbate, but may not transport significant quantities of ascorbic acid in vivo.视觉信号转导信息来源:本站原创生物谷网站The signal transduction cascade responsible for sensing light in vertebrates is one of the best studied signal transduction processes, and is initiated by rhodopsin in rod cells, a member of the G-protein coupled receptor gene family. Rhodopsin remains the only GPCR whose structure has been resolved at high resolution. Rhodopsinin the discs of rod cells contains a bound 11-cis retinal chromophore, a small molecule derived from Vitamin A that acts as the light sensitive portion of the receptor molecule, absorbing light to initiate the signal transduction cascade. When light strikes 11-cis retinal and is absorbed, it isomerizes to all-trans retinal, changing the shape of the molecule and the receptor it is bound to. This change in rhodopsin抯shape alters its interaction with transducin, the member of the G-protein gene family that is specific in its role in visual signal transduction. Activation of transducin causes its alpha subunit to dissociate from the trimer and exchange bound GDP for GTP, activating in turn a membrane-bound cyclic-GMP specific phosphodiesterase that hydrolyzes cGMP. In the resting rod cell, high levels of cGMP associate with a cyclic-GMP gated sodium channel in the plasma membrane, keeping the channels open and the membrane of the resting rod cells depolarized. This is distinct from synaptic generation of action potentials, in which stimulation induces opening of sodium channels and depolarization. When cGMP gated channels in rod cells open, both sodium and calcium ions enter the cell, hyperpolarizing the membrane and initiating the electrochemical impulse responsible for conveying the signal from the sensory neuron to the CNS. The rod cell in the resting state releases high levels of the inhibitory neurotransmitter glutamate, while the release of glutamate is repressed by the hyperpolarization in the presence of light to trigger a downstream action potential by ganglion cells that convey signals to the brain. The calcium which enters the cell also activates GCAP, which activates guanylate cyclase (GC-1 and GC-2) to rapidly produce more cGMP, ending the hyperpolarization and returning the cell to its resting depolarized state. A protein called recoverin helps mediate the inactivation of the signal transduction cascade, returning rhodopsin to its preactivated state, along with the rhodopsin kinase Grk1. Phosphorylation of rhodopsin by Grkl causes arrestin to bind, helping to terminate the receptor activation signal. Dissociation and reassociation of retinal, dephosphorylation of rhodopsin and release of arrestin all return rhodopsin to its ready state, prepared once again to respond to light.VEGF,低氧信息来源:本站原创生物谷网站Vascular endothelial growth factor (VEGF) plays a key role in physiological blood vessel formation and pathological angiogenesis such as tumor growth and ischemic diseases. Hypoxia is a potent inducer of VEGF in vitro. The increase in secreted biologically active VEGF protein from cells exposed to hypoxia is partly because of an increased transcription rate, mediated by binding of hypoxia-inducible factor-1 (HIF1) to a hypoxia responsive element in the 5'-flanking region of the VEGF gene. bHLH-PAS transcription factor that interacts with the Ah receptor nuclear translocator (Arnt), and its predicted amino acid sequence exhibits significant similarity to the hypoxia-inducible factor 1alpha (HIF1a) product. HLF mRNA expression is closely correlated with that of VEGF mRNA.. The high expression level of HLF mRNA in the O2 delivery system of developing embryos and adult organs suggests that in a normoxic state, HLF regulates gene expression of VEGF, various glycolytic enzymes, and others driven by the HRE sequence, and may be involved in development of blood vessels and the tubularsystem of lung. VEGF expression is dramatically induced by hypoxia due in large part to an increase in the stability of its mRNA. HuR binds with high affinity and specificity to the VRS element that regulates VEGF mRNA stability by hypoxia. In addition, an internal ribosome entry site (IRES) ensures efficient translation of VEGF mRNA even under hypoxia. The VHL tumor suppressor (von Hippel-Lindau) regulates also VEGF expression at a post-transcriptional level. The secreted VEGF is a major angiogenic factor that regulates multiple endothelial cell functions, including mitogenesis. Cellular and circulating levels of VEGF are elevated in hematologic malignancies and are adversely associated with prognosis. Angiogenesis is a very complex, tightly regulated, multistep process, the targeting of which may well prove useful in the creation of novel therapeutic agents. Current approaches being investigated include the inhibition of angiogenesis stimulants (e.g., VEGF), or their receptors, blockade of endothelial cell activation, inhibition of matrix metalloproteinases, and inhibition of tumor vasculature. Preclinical, phase I, and phase II studies of both monoclonal antibodies to VEGF and blockers of the VEGF receptor tyrosine kinase pathway indicate that these agents are safe and offer potential clinical utility in patients with hematologic malignancies.TSP-1诱导细胞凋亡信息来源:本站原创生物谷网站As tissues grow they require angiogenesis to occur if they are to be supplied with blood vessels and survive. Factors that inhibit angiogenesis might act as cancer therapeutics by blocking vessel formation in tumors and starving cancer cells. Thrombospondin-1 (TSP-1) is a protein that inhibits angiogenesis and slows tumor growth, apparently by inducing apoptosis of microvascular endothelial cells that line blood vessels. TSP-1 appears to produce this response by activating a signaling pathway that begins with its receptor CD36 at the cell surface of the microvascular endothelial cell. The non-receptor tyrosine kinase fyn is activated by TSP-1 through CD36, activating the apoptosis inducing proteases like caspase-3 and p38 protein kinases. p38 is a mitogen-activated kinase that also induces apoptosis in some conditions, perhaps through AP-1 activation and the activation of genes that lead to apoptosis.Trka信号转导信息来源:本站原创生物谷网站Nerve growth factor (NGF) is a neurotrophic factor that stimulates neuronal survival and growth through TrkA, a member of the trk family of tyrosine kinase receptors that also includes TrkB and TrkC. Some NGF responses are also mediated or modified by p75LNTR, a low affinity neurotrophin receptor. Binding of NGF to TrkA stimulates neuronal survival, and also proliferation. Pathways coupled to these responses are linked to TrkAthrough association of signaling factors with specific amino acids in the TrkA cytoplasmic domain. Cell survival through inhibition of apoptosis is signaled through activation of PI3-kinase and AKT. Ras-mediated signaling and phospholipase C both activate the MAP kinase pathway to stimulate proliferation.dbpb调节mRNA信息来源:本站原创生物谷网站Endothelial cells respond to treatment with the protease thrombin with increased secretion of the PDGF B-chain. This activation occurs at the transcriptional level and a thrombin response element was identified in the promoter of the PDGF B-chain gene. A transcription factor called the DNA-binding protein B (dbpB) mediates the activation of PDGF B-chain transcription in response to thrombin treatment. DbpB is a member of the Y box family of transcription factors and binds to both RNA and DNA. In the absence of thrombin, endothelial cells contain a 50 kD form of dbpB that binds RNA in the cytoplasm and may play a role as a chaperone for mRNA. The 50 kD version of dbpB also binds DNA to regulate genes containing Y box elements in their promoters. Thrombin activation results in the cleavage of dbpB to a 30 kD form. The proteolytic cleavage releases dbpB from RNA in the nucleus, allowing it to enter the nucleus and binds to a regulatory element distinct from the site recognized by the full length 50 kD dbpB. The genes activated by cleaved dbpB include the PDGF B chain. Dephosphorylation of dbpB also regulates nuclear entry and transcriptional activation.RNA digestion in vitro can release dbpB in its active form, suggesting that the protease responsible for dbpB may be closely associated in a complex. Identification of the protease that cleaves dbpB, the mechanisms of phosphorylation and dephosphorylation, and elucidation of the signaling path by which thrombin induces dbpB will provide greater understanding of this novel signaling pathway.CARM1甲基化信息来源:本站原创生物谷网站Several forms of post-translational modification regulate protein activities. Recently, protein methylation by CARM1 (coactivator-associated arginine methyltransferase 1) has been observed to play a key role in transcriptional regulation. CARM1 associates with the p160 class of transcriptional coactivators involved in gene activation by steroid hormone family receptors. CARM1 also interacts with CBP/p300 transcriptional coactivators involved in gene activation by a large variety of transcription factors, including steroid hormone receptors and CEBP. One target of CARM1 is the core histones H3 and H4, which are also targets of the histone acetylase activity of CBP/p300 coactivators. Recruitment of CARM1 to the promoter region by binding to coactivators increases histone methylation and makes promoter regions more accessible for transcription. Another target of CARM1 methylation is a coactivator it interacts with, CBP. Methylation of CBP by CARM1 blocks。
综㊀㊀述 角膜损伤修复与Notch信号通路李宗源㊀综述㊀黄一飞㊀王丽强㊀审校解放军总医院第一医学中心眼科ꎬ北京100853通信作者:王丽强ꎬEmail:liqiangw301@163.com㊀㊀ʌ摘要ɔ㊀Notch信号通路在胚胎期细胞命运转归和成体组织稳态的维持中起重要作用ꎮ各种研究已经证明Notch信号通路在角膜损伤后修复及角膜生理性稳态的维持中有重大意义ꎬ角膜缘干细胞主要通过抑制Notch信号通路来抑制细胞的分化和增生ꎻ在角膜上皮早期修复阶段生理性下调促进细胞迁移和伤口覆盖ꎬ后期修复阶段生理性上调与防止角膜上皮细胞过度分层和维持细胞分化相关ꎻ角膜基质损伤后纤维化与Notch信号通路相关ꎻ角膜内皮损伤后转化生长因子 ̄β(TGF ̄β)诱导的内皮-间质转化(EnMT)过程有Notch信号通路的直接参与ꎻ此外ꎬNotch信号通路与14 ̄3 ̄3σ㊁表皮生长因子受体(EGFR)㊁Sirt6㊁微小RNA(miRNA)㊁基质金属蛋白酶(MMPs)在角膜上皮稳态维持㊁角膜上皮分化㊁角膜基质过度炎症反应㊁角膜新生血管生成等存在相互作用ꎮ本文就Notch信号通路在角膜各层损伤修复中的功能进行综述ꎮʌ关键词ɔ㊀角膜疾病ꎻ修复ꎻ再生ꎻ信号转导ꎻNotch通路基金项目:国家重点研发计划项目(2017YFA0103200)ꎻ北京市自然科学基金项目(18L2195)DOI:10.3760/cma.j.issn.2095 ̄0160.2020.02.013CornealwoundhealingandtheNotchsignalingpathwayLiZongyuanꎬHuangYifeiꎬWangLiqiangDepartmentofOphthalmologyꎬPLAGeneralHospitalꎬBeijing100853ꎬChinaCorrespondingauthor:WangLiqiangꎬEmail:liqiangw301@163.com[Abstract]㊀TheNotchsignalingpathwayplaysanimportantroleincellfateandhomeostasis.VariousstudieshaveprovedthattheNotchsignalingpathwayhasstrongeffectsoncornealwoundhealingandthemaintenanceofcornealhomeostasis.LimbalstemcellsinhibitdifferentiationandproliferationbyinhibitingtheNotchsignalingpathway.Physiologicdownregulationpromotescellmigrationandwoundcoverageintheearlystageofcornealepithelialrepairꎬandphysiologicupregulationinthelatestageofcornealepithelialrepairisrelatedtopreventingexcessivestratificationofcornealepithelialcellsandmaintainingcelldifferentiation.FibrosisiscorrelatedwithNotchaftercornealstromalinjury.TheNotchsignalingpathwayisdirectlyinvolvedintheendothelial ̄to ̄mesenchymaltransitioninducedbytransforminggrowthfactor ̄βaftercornealendothelialinjury.InadditionꎬthereareinteractionsbetweentheNotchsignalingpathwayand14 ̄3 ̄3sigmaꎬepidermalgrowthfactorreceptorꎬSirt6ꎬmicroRNAꎬandmatrixmetalloproteinasesinmaintainingcornealepithelialhomeostasisꎬcornealepithelialdifferentiationꎬcornealstromalexcessiveinflammatoryresponseꎬcornealneovascularizationꎬetc.ThisreviewsummarizesthefunctionoftheNotchsignalingpathwayincornealwoundhealing.[Keywords]㊀CornealdiseaseꎻRepairꎻRegenerationꎻSignalingtransductionꎻNotchsignalingpathwayFundprogram:NationalKeyR&DProgramofChina(2017YFA0103200)ꎻBeijingMunicipalNaturalScienceFoundation(18L2195)DOI:10.3760/cma.j.issn.2095 ̄0160.2020.02.013㊀㊀角膜盲是全球第二大致盲疾病ꎬ而全球供体角膜的匮乏制约着角膜移植手术的普及ꎬ因此角膜损伤修复机制的研究对于发现潜在的治疗靶点尤为重要ꎮNotch信号通路是胚胎发育和组织稳态中发挥细胞间信号传递和决定细胞命运转归的一条高度保守的信号通路ꎬ是调节多种组织和细胞增生㊁分化和凋亡的关键通路[1]ꎮNotch信号通路包括经典和非经典通路[2-3]ꎮ经典Notch信号通路是由相邻细胞间的配体-受体相互作用引发的ꎬ通过Notch胞内区(NotchintracellulardomainꎬNICD)㊁RBP ̄Jκ家族核蛋白复合基因[哺乳动物的RBP ̄J㊁果蝇的Su(H)和秀丽隐杆线虫的Lag ̄1]和Mastermind活化Hes1基因ꎮ非经典Notch信号通路广义上包括不通过RBP ̄Jκ和Hes/Hey基因活化的几种Notch表达活性的模式[2]ꎮNotch信号异常与各种疾病综合征和恶性疾病相关ꎬ包括发育畸形㊁神经退行性疾病㊁代谢紊乱等ꎮ角膜由5层构成ꎬ各层来源不同ꎮ角膜上皮来源于表皮外胚层ꎬ角膜基质层和内皮层来源于神经外胚层ꎬ前弹力层由基质前层前部细纤维形成ꎬ后弹力层由内皮细胞分泌形成[4]ꎮ角膜透明ꎬ无血管ꎬ有弹性ꎬ具有较大的屈光度ꎬ表面被泪膜覆盖ꎮ角膜作为前部屏障可保护眼表免受各种来自物理㊁化学以及微生物感染的损伤ꎬ因此角膜需要强大的持续自我更新能力和快速的伤口愈合反应ꎬ这一过程需要细胞增生㊁迁移㊁分化和凋亡之间的协调作用ꎮ正常情况下角膜上皮1 5mm的损伤可在24h内愈合[5]ꎬ这种损伤后快速修复对于维持视力和角膜屏障功能至关重要ꎮ目前研究表明多种信号通路和生长因子参与角膜稳态和伤口愈合的调节[6]ꎬ但精确的分子机制仍未完全明确ꎮ本研究就角膜损伤修复过程中Notch经典通路的作用进行综述ꎬ并展望了角膜损伤修复的新靶点和新策略ꎮ1㊀Notch信号通路Notch信号通路是广泛使用的细胞间通信通路ꎬ决定着不同细胞的命运转归ꎮNotch基因最早在1917年黑腹果蝇中发现ꎬ因其功能缺失造成果蝇翅膀边缘缺刻(Notch)而命名ꎮ过去几十年的研究认为Notch信号通路是一种进化上广泛存在㊁高度保守且复杂的信号传导通路ꎮ与其他细胞间信号通路相比ꎬNotch通路有其独特性ꎮ首先ꎬ经典Notch信号以近分泌的方式传递信号ꎬ意味着信号传递发生在相邻细胞之间并且需要细胞直接接触ꎬ而大多数信号通路ꎬ如Wnt通路是依赖旁分泌传递信号ꎬ通过扩散和/或主动运输将分泌的配体运至远处靶细胞ꎮ这一独特性与经典Notch通路嵌入细胞膜中的跨膜蛋白相关ꎮ其次ꎬNotch信号通路缺乏信号放大和二次利用信号能力ꎬ因此对细胞表面传递到细胞核的信号强度非常敏感ꎬ在生理环境中Notch通路各级联信号的严格调节对最佳信号传递起到至关重要的作用ꎮ例如ꎬNotch信号通路在果蝇发育中利用侧抑制来决定细胞命运转归㊁选择细胞分化方向等[7]ꎮ另外ꎬ调节Notch信号通路可引起细胞广泛的增生㊁分化或凋亡ꎮ1.1㊀Notch信号通路的组成及激活Notch信号通路包括Notch受体㊁Notch配体㊁DNA结合蛋白(CBF ̄1SuppressorofhairlessLagꎬCSL)㊁相关调节因子和靶基因等ꎮNotch受体(Notch1㊁2㊁3和4)是由胞外区㊁跨膜区和NICD组成的一种单次跨膜糖蛋白ꎻNotch配体包括Delta ̄like家族(DLL1㊁DLL3和DLL4)㊁Jagged家族(Jagged1和Jagged2)和DSL蛋白(Delta㊁Serrate和Lag ̄2)ꎮ近期研究发现ꎬDelta ̄like配体属于频率调控ꎬDLL1触发活动脉冲信号ꎬ随后信号衰减ꎬDLL4维持Notch的长期活性[8]ꎮ转录因子CSL在Notch信号通路中识别并结合Notch诱导基因启动子特定的DNA序列ꎮNotch下游靶基因包括E/sp1㊁HES㊁HEY㊁细胞周期蛋白D1㊁NRARP和survivin等ꎮNotch信号通路激活过程为:(1)Notch蛋白经过3次剪切(S1㊁S2㊁S3)ꎬ由NICD释放入细胞质ꎻ(2)进入细胞核与转录因子CSL结合ꎬ形成NICD/CSL转录激活复合体ꎻ(3)HES㊁HEY㊁HERP等碱性-螺旋-环-螺旋转录抑制因子家族靶基因的激活ꎮ1.2㊀Notch基因在角膜中的表达目前的研究已广泛认同Notch信号通路是一条在发育过程中决定细胞命运的重要通路ꎬ在组织和细胞中参与分化㊁增生和迁移等重要功能ꎮNotch基因在角膜中研究源自小鼠皮肤Notch1条件性敲除模型中角膜上皮出现了异常增生和角质化改变ꎮ最新研究表明Notch信号通路与角膜发育㊁增生㊁分化㊁迁移㊁损伤修复等直接相关ꎮ通过逆转录聚合酶链反应分析人角膜上皮细胞Notch信号通路相关基因的表达ꎬNotch1㊁Notch2㊁DELTA1㊁Jagged1㊁Jagged2㊁HES1和HES5高表达ꎻDelta4㊁HES2㊁HEY1㊁HEY2和HEY低表达ꎬ且不能排除非上皮来源的可能ꎻ在人角膜上皮中未检测到Notch3㊁Notch4㊁Delta3和Hes3的表达[9]ꎮ在角膜不同部位Notch基因的表达也不尽相同ꎬ通过比较人角膜缘和中央角膜上皮Notch相关基因表达发现ꎬNotch1受体和Jagged1配体在角膜缘的表达高于角膜中央上皮ꎬ且主要表达于角膜上皮的基底层ꎻNotch下游靶基因HES1和HES5在中央角膜上皮的表达显著高于角膜缘ꎻNotch2㊁Delta1和Jagged2表达在两者之间差异均无统计学意义[9]ꎮ也有研究认为在成年大鼠角膜中ꎬNotch受体仅在上皮基底层表达ꎬ可能是由于一抗的选择或样品保存处理差异导致的Notch在时间和空间表达的改变[10]ꎮ2㊀角膜损伤修复与Notch信号通路角膜损伤修复是一个涉及细胞凋亡㊁迁移㊁增生㊁分化和细胞外基质(excetralcellularmatrixꎬECM)重塑的复杂过程[11]ꎮ角膜上皮细胞㊁角膜基质细胞㊁角膜神经和免疫细胞之间的自分泌和旁分泌细胞因子介导的固有级联反应控制着这一过程[12]ꎮ角膜上皮细胞㊁基质细胞和内皮细胞的损伤修复过程有一定相似性ꎬ如3种不同层细胞修复均与细胞迁移相关ꎬ依赖生长因子和ECM的重塑ꎮ角膜上皮与角膜基质修复之间还存在交叉影响ꎬ如基质细胞生成的基质细胞生长因子(keratinocytegrowthfactorꎬKGF)和肝细胞生长因子(hepatocytegrowthfactorꎬHGF)影响上皮修复ꎻ上皮细胞分泌的白细胞介素 ̄1(interleukin ̄1ꎬIL ̄1)和血小板衍生生长因子(plateletderivedgrowthfactorꎬPDGF)调节基质损伤后反应ꎻ胰岛素样生长因子(insulinlikegrowthfactorsꎬIGF)和转化生长因子β(transforminggrowthfactor ̄βꎬTGF ̄β)可调节上皮细胞和基质细胞向肌成纤维细胞转化等[6]ꎮ除此之外ꎬ角膜各层细胞还存在特异性差异ꎮ2.1㊀角膜上皮损伤修复2.1.1㊀角膜上皮修复的生理过程㊀角膜上皮是一种外胚层衍生㊁无角质化㊁有独特角蛋白ꎬ有自我更新能力的组织ꎬ厚度为48~53μm[13]ꎮ角膜上皮是由一系列紧密连接细胞组成的屏障ꎬ厚7~8层ꎬ包括微绒毛㊁表层细胞㊁翼状细胞㊁基底细胞等附着在基底膜ꎮ细胞间通过桥粒彼此连接ꎬ通过缝隙连接在层与层之间传递信号ꎮ角膜上皮作为最外层ꎬ易于受伤且需要快速修复以防止更深层感染ꎮ角膜上皮修复是通过位于角膜缘的角膜缘干细胞(limbalstemcellsꎬLSCs)功能来实现的ꎬ解剖学上角膜缘为LSCs提供了独特的角膜缘微环境ꎮ功能上ꎬ角膜缘栅栏结构被血管网包围ꎬ血管网可阻断T淋巴细胞和抗原递呈细胞浸润ꎬ也可为LSCs提供充足营养和氧气ꎮ目前关于角膜上皮损伤修复的主流假说包括角膜缘上皮干细胞(corneallimbalepithelialstemcellsꎬLESCs)假说和角膜上皮干细胞假说ꎮ临床研究中发现ꎬ在角膜缘干细胞缺陷的情况下ꎬ单纯的穿透角膜移植必将失败[14]ꎬ表明LESCs对于角膜上皮的长期再生具有重要作用ꎮ因此LESCs假说仍是解释角膜上皮稳态和损伤修复较合理的机制ꎮ角膜上皮损伤后修复很大程度上取决于角膜缘干细胞和基底膜重塑ꎬ包括初始潜伏期和闭合期2个动力学阶段[15]ꎮ初始阶段无细胞运动ꎬ细胞数量也无明显变化ꎬ但代谢活动增加ꎬ可观察到细胞微结构重组ꎮ这一阶段伴随细胞骨架蛋白和整合蛋白ꎬ如α6和β4的合成增加[16]ꎮ闭合阶段包括细胞迁移㊁增生和分化ꎮ细胞迁移是伤口周边细胞迁移后覆盖缺损区域ꎻ细胞增生㊁分化是指上皮细胞经这2个过程后通过分层恢复原始复层结构ꎬ并与基底建立连接ꎮ上述阶段并非单一有序进行ꎬ覆盖受伤区域㊁恢复细胞密度和细胞附着建立是同步的ꎮ当角膜上皮再生机制失效时ꎬ会发生角膜上皮持续缺损ꎮ2.1.2㊀角膜上皮的损伤修复与Notch信号通路㊀Notch信号通路与角膜上皮损伤修复有密切联系ꎬ但Notch影响上皮细胞增生的机制至今仍不明ꎮNotch基因功能具有双向性ꎬ一方面增强Notch活性可刺激上皮细胞增生ꎬ从而有效地促进角膜创伤愈合ꎻ另一方面Notch可以抑制角膜上皮增生ꎮ这种特性可能与Notch信号通路所在部位和信号强度相关ꎮ生理状态下ꎬNICD的过表达不会引起角膜上皮细胞增生和分化异常ꎬ但增强Notch1活性可在损伤后早期刺激角膜上皮细胞增生ꎬ通过改变角膜上皮动力学有效促进角膜创伤愈合ꎮ在损伤修复中增加Notch1活性有助于角膜上皮细胞增生ꎬ这可能是某些创伤反应因子提高了靶细胞对Notch信号的敏感性[5]ꎮ在体内损伤模型中ꎬHes1的表达与细胞增生活性呈正相关ꎬ而在体外三维培养模型中Hes1的过表达却抑制细胞增生[17]ꎮ这种看似矛盾的现象正是Notch通路双向性的体现ꎮ通过进一步建立体内和体外Notch抑制模型ꎬ发现抑制Notch通路加速角膜上皮细胞迁移和伤口闭合ꎬ但这一过程与细胞迁移相关ꎬ与细胞增生无关ꎮ此外ꎬ抑制Notch信号通路还将降低细胞-基质黏附和肌动蛋白细胞骨架改变ꎬ这些都与促进细胞迁移相关[5]ꎮNotch1基因敲除小鼠(Notch1-/-)角膜上皮损伤后细胞再生延迟㊁紧密连接形成延迟且无法恢复角膜上皮的屏障功能ꎮ除此之外ꎬNotch1-/-小鼠出生后4~6周ꎬ角膜基质开始混浊ꎬ组织病理学检查提示有明显的炎症㊁溃疡和新生血管形成[18-19]ꎮNotch信号通路在不同损伤修复阶段表现不同ꎮNotch1表达水平在角膜损伤早期明显下调ꎬ在损伤后期恢复ꎮ角膜伤口愈合早期阶段(4h)ꎬ抑制Notch信号通路对伤口周边上皮细胞形态和层数无显著影响ꎮ在上皮损伤修复的后期阶段(24h)ꎬ抑制Notch信号通路则显著促进上皮细胞分层和再上皮化ꎮ由此可见ꎬNotch信号通路在角膜上皮早期修复阶段生理性下调会促进细胞迁移和伤口覆盖ꎬ在后期修复阶段生理性上调可能与防止角膜上皮细胞过度分层和维持细胞的分化相关ꎮ在角膜上皮损伤修复过程中ꎬNotch通路与其他基因和小分子化合物具有联系和交叉作用ꎮ(1)14 ̄3 ̄3σ与P63㊀14 ̄3 ̄3σ是stratifin蛋白家族中一员ꎬ特异性表达在分化的鳞状上皮中ꎬ在细胞信号转导和细胞周期调节中起重要作用ꎬ与皮肤上皮分层和分化相关[20]ꎮ在表皮基底祖细胞中14 ̄3 ̄3σ表达被p63抑制ꎬ当表皮祖细胞分化时p63表达减少ꎬ进而诱导14 ̄3 ̄3σ表达增加ꎮ通过对14 ̄3 ̄3σ突变Er小鼠模型的研究ꎬ确定了14 ̄3 ̄3σ在调控表皮上皮发育和分化中的作用[21]ꎮ14 ̄3 ̄3σ是角膜上皮细胞稳态的关键调节因子ꎬ对于调控角膜上皮细胞增生和分化是必需的ꎮ当14 ̄3 ̄3σ活性被抑制时ꎬNotch1和Notch2转录减少ꎬ同时出现上皮细胞增生和分化异常ꎻ当以过表达方式恢复Notch活性时ꎬ上皮细胞增生和分化异常均得到改善[22]ꎮ由此说明在角膜上皮干前体细胞分化过程中ꎬ14 ̄3 ̄3σ调控上游Notch1的表达ꎬ在调节角膜上皮细胞分化中起重要作用[23]ꎮ(2)表皮生长因子受体(epidermalgrowthfactorreceptorꎬEGFR)与p53㊀EGFR是表皮生长因子受体家族成员之一ꎬ通过下调p53基因转录负调控Notch1基因表达ꎬ抑制EGFR信号可增强Notch通路并增强上皮细胞分化ꎮ若改变角膜上皮EGFR通路ꎬ将导致细胞凋亡增加㊁细胞增生减少ꎬ伤口愈合延迟[24]ꎮ(3)Sirt6㊀组蛋白去乙酰化酶Sirt6是角膜炎症反应的关键调控因子ꎬ与角膜上皮损伤修复㊁维持上皮完整性和角膜透明度相关ꎮSirt6缺乏将导致角膜上皮伤口不完全愈合和愈合时间延迟ꎻ还与角膜基质的过度炎症反应和Notch通路异常相关ꎬ这些最终导致角膜上皮角质化和角膜混浊的发生[25]ꎮ(4)微小RNA(microRNAꎬmiRNA)㊀miRNA在干细胞的维持和分化中发挥作用ꎮ角膜上皮以丰富的糖原储备为主要能量来源ꎮmiR ̄31通过负调控抑制缺氧诱导因子(factorinhibitinghypoxiainduciblefactor ̄1ꎬFIH ̄1)维持角膜上皮细胞中糖原的代谢[26]ꎮNotch通路部分通过miR ̄31/FIH ̄1来影响上皮细胞分化ꎬFIH ̄1对Notch羟基化ꎬ下调Notch通路ꎬ负调节角膜上皮细胞分化[27]ꎮ由于FIH ̄1蛋白在角膜缘上皮细胞中大量存在ꎬ但在角膜上皮细胞中几乎不存在ꎬ还可推断在角膜缘中低水平miR ̄31和高水平FIH ̄1(其减弱Notch通路信号)可能是维持角膜缘上皮祖细胞未分化表型的一种机制[27]ꎮmiR ̄184参与细胞发育㊁增生和迁移等重要过程ꎮmiR ̄184的不同突变与晶状体㊁角膜营养不良和家族性圆锥角膜等相关ꎮ最近研究表明ꎬmiR ̄184在角膜上皮细胞中通过调节Notch和p63之间的平衡ꎬ在Notch通路及其上游抑制上皮细胞的增生作用ꎬ也可直接抑制K15和FIH1加强Notch依赖性的上皮细胞分化作用[28]ꎮ(5)基质金属蛋白酶(matrixmetalloproteinaseꎬMMPs)㊀MMPs是一种维持ECM重塑和血管生成平衡的关键因子ꎮ除了参与形成ECM外ꎬ角膜在修复过程中还会合成多种MMPsꎬ包括MMP ̄1㊁MMP ̄2㊁MMP ̄3㊁MMP ̄9和MMP ̄14[29]ꎮMMP ̄12通过增强上皮细胞迁移和中性粒细胞浸润促进角膜上皮损伤后早期修复过程ꎬ在涉及角膜全层损伤的化学损伤模型中ꎬ对预防角膜纤维化也具有积极作用[30]ꎮMMP ̄14是一种跨膜MMPꎬ主要参与ECM蛋白水解㊁外泌体运输和细胞迁移等功能ꎬ对角膜新生血管的生成至关重要ꎮ在小鼠角膜受损模型中MMP ̄14的过度表达可降低Ⅲ型胶原和α平滑肌肌动蛋白(α ̄smoothmuscleactinꎬα ̄SMA)的表达来预防角膜纤维化ꎬ维持角膜的透明性[29]ꎮMMP ̄14对Notch信号有负调控作用ꎬMMP ̄14通过切割Notch配体DLL1ꎬ抑制Notch1信号ꎻ抑制MMP ̄14增强了Notch信号ꎬ表明MMP ̄14与Notch之间存在负反馈调节通路ꎮ2.1.3㊀角膜上皮损伤修复与角膜基质的联系㊀角膜基质细胞表达许多对上皮细胞具有特异性作用的生长因子和细胞因子ꎬ如表皮生长因子(epidermalgrowthfactorꎬEGF)㊁PDGF㊁TGF ̄α㊁TGF ̄β㊁成纤维细胞生长因子1(fibroblastgrowthfactor ̄1ꎬFGF ̄1)㊁FGF ̄2㊁IGF ̄I㊁KGF㊁HGF㊁胸腺素㊁IL ̄1㊁IL ̄6㊁IL ̄10ꎬ以及肿瘤坏死因子αꎬ这些因子也与角膜上皮损伤修复密切相关[11]ꎮ这些小分子是角膜上皮细胞的强有丝分裂原ꎬ临床上也以滴眼液或药物与角膜接触镜相结合的形式局部促进角膜上皮细胞增生ꎬ加速上皮伤口愈合[12]ꎮ2.2㊀角膜基质的损伤修复角膜基质细胞和角膜成纤维细胞产生大量的ECM成分ꎬ如巢蛋白 ̄1㊁巢蛋白 ̄2和基底膜蛋白ꎬ这些成分对于正常生理状态维持和基质完全再生是必要的[31]ꎮ角膜基质损伤后修复通常取决于损伤类型和程度ꎮ角膜基质损伤后首要变化是损伤部位周围基质细胞群凋亡ꎬ细胞凋亡是由细胞因子ꎬ如上皮细胞分泌的IL ̄1诱导ꎮ角膜基质修复早期阶段ꎬ伤口周围基质细胞活化为成纤维细胞ꎬ重新获得迁移能力并进入细胞周期ꎬ这些损伤后产生的成纤维细胞分泌的ECM不同于正常基质细胞ꎬ如肌腱蛋白和纤维连接蛋白仅在修复组织中合成[32]ꎮ这些成纤维细胞通过肌动蛋白细胞骨架重塑ꎬ从星状变为细长状并获得应力ꎮ成纤维细胞还将下调基质细胞蛋白的表达ꎬ并分泌重塑ECM所需的蛋白酶ꎬ主要是MMP[32]ꎮ到达损伤部位后ꎬ成纤维细胞开始表达α ̄SMA和肌间线蛋白并上调波形蛋白表达ꎬ最终转化为介导伤口闭合具有高度运动能力的肌成纤维细胞[33]ꎮ肌成纤维细胞的角膜晶体蛋白表达减少ꎬ分泌异常ECMꎬ是导致角膜基质纤维化和角膜混浊的主要病理过程[34]ꎮ一旦肌成纤维细胞介导的基质伤口愈合后ꎬ基质中肌成纤维细胞数量会缓慢减少ꎮ在受伤数月或数年后ꎬ这些肌成纤维细胞及其产生的混浊基质在上皮基底膜(epithelialbasementmembraneꎬEBM)完全再生后ꎬ被规则的角膜ECM缓慢取代[35]ꎬ逐渐恢复角膜透明性ꎮ这一过程中肌成纤维细胞和基质细胞相互竞争ꎬ最终角膜基质细胞将吸收杂乱无序的细胞外基质并重新组成前基质[36]ꎮEBM的再生是决定角膜基质是否完全再生的关键因素ꎬ包括透明性和视力恢复[36]ꎮ再生EBM的异常结构和功能使上皮来源的生长因子ꎬ如PDGF和TGF ̄β(只有TGF ̄β1和TGF ̄β2在这个过程中具有活性[37]ꎬTGF ̄β3具有抗瘢痕形成的效果[38-39])长期进入角膜基质ꎬ这两者在前基质细胞中持续存在ꎬ诱导前体细胞分化为肌成纤维细胞[11]ꎮ根据目前的研究认为ꎬNotch信号可能与角膜基质损伤后纤维化相关ꎬ但总体看Notch信号在角膜基质细胞损伤修复过程中调节能力有限[10]ꎮ2.3㊀角膜内皮的损伤修复2.3.1㊀角膜内皮修复的生理过程㊀角膜内皮由单层大小均匀㊁低阻力紧密连接的六边形细胞组成ꎮ角膜内皮细胞维持正常形态特征ꎬ保持完整紧密连接和正常细胞密度ꎬ对于保持角膜脱水化㊁透明和维持视力至关重要[40]ꎮ成人角膜内皮细胞(cornealendothelialcellsꎬCECs)在体内具有低丝分裂活性ꎬ原因是缺乏有丝分裂刺激ꎬ细胞接触抑制和房水中抗有丝分裂因子存在ꎮ角膜内皮细胞ꎬ特别是人角膜内皮细胞ꎬ增生能力差ꎬ体外难以培养ꎬ易发生内皮间质转化(endothelialtomesenchymaltransitionꎬEnMT)和细胞衰老[41]ꎬ近年研究表明ROCK抑制剂Y ̄27632对于包括兔㊁灵长类和人的角膜内皮细胞的迁移㊁黏附起增强作用[42-43]ꎮ角膜内皮损伤修复与上皮和基质显著不同ꎮ内皮损伤后不同动物的修复方式不同ꎬ在兔中损伤周围的内皮细胞通过细胞移行和有丝分裂修复伤口ꎬ在成人角膜内皮受伤后仅依靠角膜缘内皮细胞向中心移行和损伤周围细胞扩大愈合伤口ꎬ当角膜内皮密度<500/mm2时会发生角膜内皮失代偿ꎬ出现大泡性角膜病变[40]ꎮ近些年研究表明ꎬCECs在受到化学㊁机械或其他损伤后发生EnMTꎬ主要通过TGF ̄β介导细胞迁移和扩大愈合伤口ꎮEnMT包括内皮细胞形态特征和分泌ECM改变ꎮECM改变包括钙黏蛋白下调ꎬⅣ型胶原基底膜转变为Ⅰ型胶原基底膜和肌成纤维细胞引起的α ̄SMA表达增加等ꎮ已知作为有丝分裂原影响内皮细胞移行以促进内皮迁移和伤口愈合的生长因子包括EGF㊁PDGF ̄BB和FGF ̄2等ꎮ此外ꎬIL ̄1β和血管内皮生长因子(vascularendothelialgrowthfactorꎬVEGF)也促进内皮细胞迁移ꎻIGF ̄Ⅰ和IGF ̄Ⅱ对细胞迁移无明显影响ꎬIL ̄4会降低细胞的迁移能力ꎮ2.3.2㊀角膜EnMT与Notch信号通路㊀近期研究表明胚胎时期Notch信号通路在有效区域激活EnMT[44]ꎬNotch信号通路激活后内皮细胞发生与EnMT相一致的形态学和功能改变ꎮ体外培养人内皮细胞第1代可检测Jagged1㊁Jagged2㊁Notch1㊁Notch2和Hes1等Notch相关基因表达ꎬ并在后续代数中更为显著ꎬNotch抑制剂DA几乎完全阻断TGF ̄β诱导的EnMT也证实Notch通路位于TGF ̄β信号下游的观点ꎮTGF ̄β诱导在CECs的EnMT和角膜后纤维膜(retrocornealfibrousmembraneꎬRCFM)形成中起重要作用ꎮ抑制这一靶点可预防损伤性角膜内皮纤维化ꎮ在体外培养内皮细胞过程中ꎬVEGF可诱导Notch1受体和Notch配体DLL4的表达ꎮNotch通过上调MMP ̄9和MMP ̄14在VEGF下游起作用ꎬMMP ̄14通过切割Notch配体DII1ꎬ负调节Notch信号传导[29]ꎮ在体冻伤模型中ꎬ局部应用DAPT可显著减轻角膜水肿ꎬ提高透明度ꎬ后期RCFM的形成明显减少ꎬ可能与DAPT抑制EnMTꎬCECs密度升高改善角膜内皮功能相关[45]ꎮ以上体内体外均证实了EnMT与Notch信号通路的相关性ꎮ由于角膜植片来源匮乏ꎬ通过抑制Notch信号通路减轻CECs的EnMT可为组织工程角膜内皮植片提供可靠的内皮种子细胞ꎬ也为预防内皮损伤后RCFM形成提供新的治疗思路ꎮ3 展望Notch信号通路调节全身各器官组织发育和出生后细胞命运转归以及维持体内稳态起到至关重要作用ꎮNotch信号通路在不同时间㊁不同组织㊁不同水平中起作用ꎮ此外ꎬNotch信号通路在调节细胞命运上存在物种和组织特异性差异ꎮ例如Notch信号通路在肌肉祖细胞自我更新㊁胚胎发育分化和成年组织稳态中发挥不同功能[46]ꎻ在调节视网膜前体细胞分化为视网膜神经节细胞过程中起着重要的作用[47]ꎻ在角膜损伤修复不同阶段Notch信号通路相关蛋白表达量㊁表达部位和功能均不同[11ꎬ35ꎬ48]ꎮ由于Notch通路调节 双向性 ꎬ时间和信号强度在不同组织稳态维持中具有特异性ꎮ同一组织中ꎬNotch又与p63㊁p53㊁14 ̄3 ̄3σ㊁EGFR㊁Sirt6㊁miRNA㊁MMPs等基因或小分子化合物存在交叉作用ꎮ由此可见ꎬNotch信号通路对于机体组织的调控远比我们目前认识的复杂ꎮXiang等[49]报道了Notch抑制剂DAPT与其他4种小分子化合物联用(腺苷酸环化酶激活剂FSK㊁TGF ̄β抑制剂SB43㊁Wnt抑制剂IWP2和BMP抑制剂LDN193189)实现了体外抑制EMT并维持终末分化细胞特性ꎮ在角膜研究中发现体外大鼠LESCs主要通过抑制Notch信号通路抑制细胞的分化和增生[50]ꎮ以上为角膜损伤修复再生提供了新的思路ꎬ多条通路的靶点共同干预是否比单一通路靶点进行干预更有效促进组织损伤后修复和再生仍不清楚ꎮ在未来研究中ꎬ可以将Notch通路和其他经典通路的交互作用作为研究重点ꎬ研究多通路协同修复损伤的机制以发现新的潜在治疗靶点ꎬ为预防和治疗角膜盲提供新的策略ꎮ利益冲突㊀所有作者均声明不存在利益冲突参考文献[1]BraySJ.Notchsignalling:asimplepathwaybecomescomplex[J].NatRevMolCellBiolꎬ2006ꎬ7(9)ʒ678-689.DOI:10.1038/nrm2009. [2]KopanRꎬIlaganMX.ThecanonicalNotchsignalingpathway:unfoldingtheactivationmechanism[J].Cellꎬ2009ꎬ137(2)ʒ216-233.DOI:10.1016/j.cell.2009.03.045.[3]SanalkumarRꎬDhaneshSBꎬJamesJ.Non ̄canonicalactivationofNotchsignaling/targetgenesinvertebrates[J].CellMolLifeSciꎬ2010ꎬ67(17)ʒ2957-2968.DOI:10.1007/s00018 ̄010 ̄0391 ̄x. [4]GrawJ.Thegeneticandmolecularbasisofcongenitaleyedefects[J].NatRevGenetꎬ2003ꎬ4(11)ʒ876-888.DOI:10.1038/nrg1202. [5]LuHꎬLuQꎬZhengYꎬetal.Notchsignalingpromotesthecornealepitheliumwoundhealing[J].MolVisꎬ2012ꎬ18ʒ403-411. [6]YuFSꎬYinJꎬXuKꎬetal.Growthfactorsandcornealepithelialwoundhealing[J].BrainResBullꎬ2010ꎬ81(2-3)ʒ229-235.DOI:10.1016/j.brainresbull.2009.08.024.[7]LaydenMJꎬMartindaleMQ.Non ̄canonicalNotchsignalingrepresentsanancestralmechanismtoregulateneuraldifferentiation[J/OL].Evodevoꎬ2014ꎬ5ʒ30[2019-02-01].https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4335385/.DOI:10.1186/2041 ̄9139 ̄5 ̄30. [8]MegasonSG.Dynamicencodinginthenotchpathway[J].DevCellꎬ2018ꎬ44(4)ʒ411-412.DOI:10.1016/j.devcel.2018.02.006. [9]DjalilianARꎬNamavariAꎬItoAꎬetal.Down ̄regulationofNotchsignalingduringcornealepithelialproliferation[J].MolVisꎬ2008ꎬ14ʒ1041-1049.[10]MaAꎬZhaoBꎬBoultonMꎬetal.AroleforNotchsignalingincornealwoundhealing[J].WoundRepairRegenꎬ2011ꎬ19(1)ʒ98-106.DOI:10.1111/j.1524 ̄475X.2010.00648.x.[11]LjubimovAVꎬSaghizadehM.Progressincornealwoundhealing[J].ProgRetinEyeResꎬ2015ꎬ49ʒ17-45.DOI:10.1016/j.preteyeres.2015.07.002.[12]ZiaeiMꎬGreeneCꎬGreenCR.Woundhealingintheeye:Therapeuticprospects[J].AdvDrugDelivRevꎬ2018ꎬ126ʒ162-176.DOI:10.1016/j.addr.2018.01.006.[13]OieYꎬNishidaK.Regenerativemedicineforthecornea[J/OL].BiomedResIntꎬ2013ꎬ2013ʒ428247[2019-03-01].https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3876767/.DOI:10.1155/2013/428247.[14]LangSJꎬBöhringerDꎬGeerlingGꎬetal.Long ̄termresultsofallogenicpenetratinglimbo ̄keratoplasty:20yearsofexperience[J].Eye(Lond)ꎬ2017ꎬ31(3)ʒ372-378.DOI:10.1038/eye.2016.217. [15]LiuCYꎬKaoWW.Cornealepithelialwoundhealing[J].ProgMolBiolTranslSciꎬ2015ꎬ134ʒ61-71.DOI:10.1016/bs.pmbts.2015.05.002.[16]HynesRO.Integrins:bidirectionalꎬallostericsignalingmachines[J].Cellꎬ2002ꎬ110(6)ʒ673-687.DOI:10.1016/s0092 ̄8674(02)00971 ̄6. [17]NakamuraTꎬOhtsukaTꎬSekiyamaEꎬetal.Hes1regulatescornealdevelopmentandthefunctionofcornealepithelialstem/progenitorcells[J].StemCellsꎬ2008ꎬ26(5)ʒ1265-1274.DOI:10.1634/stemcells.2007 ̄1067.[18]VauclairSꎬMajoFꎬDurhamADꎬetal.CornealepithelialcellfateismaintainedduringrepairbyNotch1signalingviatheregulationofvitaminAmetabolism[J].DevCellꎬ2007ꎬ13(2)ʒ242-253.DOI:10.1016/j.devcel.2007.06.012.[19]MovahedanAꎬAfsharkhamsehNꎬSaghaHMꎬetal.LossofNotch1disruptsthebarrierrepairinthecornealepithelium[J/OL].PLoSOneꎬ2013ꎬ8(7)ʒe69113[2019-02-13].https://www.ncbi.nlm.nih.gov/pubmed/23874882.DOI:10.1371/journal.pone.0069113. [20]PellegriniGꎬDellambraEꎬGolisanoOꎬetal.p63identifieskeratinocytestemcells[J].ProcNatlAcadSciUSAꎬ2001ꎬ98(6)ʒ3156-3161.DOI:10.1073/pnas.061032098.[21]LiQꎬLuQꎬEstepaGꎬetal.Identificationof14 ̄3 ̄3sigmamutationcausingcutaneousabnormalityinrepeated ̄epilationmutantmouse[J].ProcNatlAcadSciUSAꎬ2005ꎬ102(44)ʒ15977-15982.DOI:10.1073/pnas.0508310102.[22]LuQꎬXinYꎬYeFꎬetal.14 ̄3 ̄3σcontrolscornealepitheliumhomeostasisandwoundhealing[J].InvestOphthalmolVisSciꎬ2011ꎬ52(5)ʒ2389-2396.DOI:10.1167/iovs.09 ̄4981.[23]XinYꎬLuQꎬLiQ.14 ̄3 ̄3sigmacontrolscornealepithelialcellproliferationanddifferentiationthroughtheNotchsignalingpathway[J].BiochemBiophysResCommunꎬ2010ꎬ392(4)ʒ593-598.DOI:10.1016/j.bbrc.2010.01.084.[24]XuKꎬYuFS.ImpairedepithelialwoundhealingandEGFRsignalingpathwaysinthecorneasofdiabeticrats[J].InvestOphthalmolVisSciꎬ2011ꎬ52(6)ʒ3301-3308.DOI:10.1167/iovs.10 ̄5670.[25]HuXꎬZhuSꎬLiuRꎬetal.Sirt6deficiencyimpairscornealepithelialwoundhealing[J].Aging(AlbanyNY)ꎬ2018ꎬ10(8)ʒ1932-1946.DOI:10.18632/aging.101513.[26]PengHꎬHamanakaRBꎬKatsnelsonJꎬetal.MicroRNA ̄31targetsFIH ̄1topositivelyregulatecornealepithelialglycogenmetabolism[J].FASEBJꎬ2012ꎬ26(8)ʒ3140-3147.DOI:10.1096/fj.11 ̄198515. [27]PengHꎬKaplanNꎬHamanakaRBꎬetal.microRNA ̄31/factor ̄inhibitinghypoxia ̄induciblefactor1nexusregulateskeratinocytedifferentiation[J].ProcNatlAcadSciUSAꎬ2012ꎬ109(35)ʒ14030-14034.DOI:10.1073/pnas.1111292109.[28]NagosaSꎬLeeschFꎬPutinDꎬetal.microRNA ̄184inducesacommitmentswitchtoepidermaldifferentiation[J].StemCellReportsꎬ2017ꎬ9(6)ʒ1991-2004.DOI:10.1016/j.stemcr.2017.10.030. [29]ChangJHꎬHuangYHꎬCunninghamCMꎬetal.Matrixmetalloproteinase14modulatessignaltransductionandangiogenesisinthecornea[J].SurvOphthalmolꎬ2016ꎬ61(4)ʒ478-497.DOI:10.1016/j.survophthal.2015.11.006.[30]WolfMꎬMaltsevaIꎬClaySMꎬetal.EffectsofMMP12oncellmotilityandinflammationduringcornealepithelialrepair[J].ExpEyeResꎬ2017ꎬ160ʒ11-20.DOI:10.1016/j.exer.2017.04.007.[31]WilsonSE.Cornealmyofibroblastbiologyandpathobiology:generationꎬpersistenceꎬandtransparency[J].ExpEyeResꎬ2012ꎬ99ʒ78-88.DOI:10.1016/j.exer.2012.03.018.[32]West ̄MaysJAꎬDwivediDJ.Thekeratocyte:cornealstromalcellwithvariablerepairphenotypes[J].IntJBiochemCellBiolꎬ2006ꎬ38(10)ʒ1625-1631.DOI:10.1016/j.biocel.2006.03.010.[33]SinghVꎬJainiRꎬTorricelliAAꎬetal.TGFβandPDGF ̄Bsignalingblockadeinhibitsmyofibroblastdevelopmentfrombothbonemarrow ̄derivedandkeratocyte ̄derivedprecursorcellsinvivo[J].ExpEyeResꎬ2014ꎬ121ʒ35-40.DOI:10.1016/j.exer.2014.02.013. [34]TorricelliAAꎬSinghVꎬAgrawalVꎬetal.Transmissionelectronmicroscopyanalysisofepithelialbasementmembranerepairinrabbitcorneaswithhaze[J].InvestOphthalmolVisSciꎬ2013ꎬ54(6)ʒ4026-4033.DOI:10.1167/iovs.13 ̄12106.[35]MimuraTꎬYamagamiSꎬAmanoS.Cornealendothelialregenerationandtissueengineering[J].ProgRetinEyeResꎬ2013ꎬ35ʒ1-17.DOI:10.1016/j.preteyeres.2013.01.003.[36]TorricelliAAꎬSanthanamAꎬWuJꎬetal.Thecornealfibrosisresponsetoepithelial ̄stromalinjury[J].ExpEyeResꎬ2016ꎬ142ʒ110-118.DOI:10.1016/j.exer.2014.09.012.[37]GuoXꎬHutcheonAEꎬZieskeJD.MolecularinsightsontheeffectofTGF ̄β1/ ̄β3inhumancornealfibroblasts[J].ExpEyeResꎬ2016ꎬ146ʒ233-241.DOI:10.1016/j.exer.2016.03.011.[38]SinghVꎬBarbosaFLꎬTorricelliAAꎬetal.Transforminggrowthfactorβandplatelet ̄derivedgrowthfactormodulationofmyofibroblastdevelopmentfromcornealfibroblastsinvitro[J].ExpEyeResꎬ2014ꎬ120ʒ152-160.DOI:10.1016/j.exer.2014.01.003.[39]KaramichosDꎬHutcheonAEꎬZieskeJD.ReversaloffibrosisbyTGF ̄β3ina3Dinvitromodel[J].ExpEyeResꎬ2014ꎬ124ʒ31-36.DOI10.1016/j.exer.2014.04.020.[40]DelMonteDWꎬKimT.Anatomyandphysiologyofthecornea[J].JCataractRefractSurgꎬ2011ꎬ37(3)ʒ588-598.DOI:10.1016/j.jcrs.2010.12.037.[41]HongoAꎬOkumuraNꎬNakaharaMꎬetal.Theeffectofap38mitogen ̄activatedproteinkinaseinhibitoroncellularsenescenceofcultivatedhumancornealendothelialcells[J].InvestOphthalmolVisSciꎬ2017ꎬ58(9)ʒ3325-3334.DOI:10.1167/iovs.16 ̄21170.[42]OkumuraNꎬUenoMꎬKoizumiNꎬetal.EnhancementonprimatecornealendothelialcellsurvivalinvitrobyaROCKinhibitor[J].InvestOphthalmolVisSciꎬ2009ꎬ50(8)ʒ3680-3687.DOI:10.1167/iovs.08 ̄2634.[43]KinoshitaSꎬKoizumiNꎬUenoMꎬetal.InjectionofculturedcellswithaROCKinhibitorforbullouskeratopathy[J].NEnglJMedꎬ2018ꎬ378(11)ʒ995-1003.DOI:10.1056/NEJMoa1712770.[44]LeongKGꎬNiessenKꎬKulicIꎬetal.Jagged1 ̄mediatedNotchactivationinducesepithelial ̄to ̄mesenchymaltransitionthroughSlug ̄inducedrepressionofE ̄cadherin[J].JExpMedꎬ2007ꎬ204(12)ʒ2935-2948.DOI:10.1084/jem.20071082.[45]LiCꎬDongFꎬJiaYꎬetal.Notchsignalregulatescornealendothelial ̄to ̄mesenchymaltransition[J].AmJPatholꎬ2013ꎬ183(3)ʒ786-795.DOI:10.1016/j.ajpath.2013.05.025.[46]BröhlDꎬVasyutinaEꎬCzajkowskiMTꎬetal.ColonizationofthesatellitecellnichebyskeletalmuscleprogenitorcellsdependsonNotchsignals[J].DevCellꎬ2012ꎬ23(3)ʒ469-481.DOI:10.1016/j.devcel.2012.07.014.[47]王迎彬ꎬ朱益华ꎬ徐国兴.Notch信号通路与视网膜神经节细胞的发育[J].中华实验眼科杂志ꎬ2007ꎬ25(10)ʒ797-800.WangYBꎬZhuYHꎬXuGX.Notchsignalingpathwayanddevelopmentofretinalganglioncells[J].ChinJExpOphthalmolꎬ2007ꎬ25(10)ʒ797-800.[48]BukowieckiAꎬHosDꎬCursiefenCꎬetal.Wound ̄healingstudiesincorneaandskin:parallelsꎬdifferencesandopportunities[J/OL].IntJMolSciꎬ2017ꎬ18(6)ʒ1257[2019-02-20].https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5486079/.DOI:10.3390/ijms18061257. [49]XiangCꎬDuYꎬMengGꎬetal.Long ̄termfunctionalmaintenanceofprimaryhumanhepatocytesinvitro[J].Scienceꎬ2019ꎬ364(6438)ʒ399-402.DOI:10.1126/science.aau7307.[50]LiJꎬChenSYꎬZhaoXYꎬetal.Ratlimbalnichecellspreventepithelialstem/progenitorcellsfromdifferentiationandproliferationbyinhibitingnotchsignalingpathwayinvitro[J].InvestOphthalmolVisSciꎬ2017ꎬ58(7)ʒ2968-2976.DOI:10.1167/iovs.16 ̄20642.(收稿日期:2019-09-22㊀修回日期:2019-11-30)(本文编辑:刘艳)读者 作者 编者本刊稿件处理流程㊀㊀本刊实行以同行审稿为基础的三级审理制度(编辑初审㊁专家外审㊁编委会终审)稿件评审ꎮ编辑部在稿件审理过程中坚持客观㊁公平㊁公正的原则ꎬ郑重承诺审稿过程中尊重和保护审稿专家㊁作者及稿件的私密权ꎮ专家审理认为不宜刊用的稿件ꎬ编辑部将告知作者专家的审理意见ꎬ对稿件处理有不同看法的作者有权向编辑部申请复议ꎬ但请写出申请理由和意见ꎮ稿件审理过程中作者可通过 中华医学会杂志社远程稿件管理系统 查询稿件的审理结果ꎮ作者如需要采用通知或退稿通知可与编辑部联系ꎮ编辑部发给作者修改再审的稿件ꎬ如2个月没有修回ꎬ视为作者自行撤稿ꎮ编辑部的各种通知将通过Email发出ꎬ投稿后和稿件审理期间请作者留意自己的电子信箱ꎮ作者自收到采用通知之日起ꎬ即视为双方建立合约关系ꎬ作者如撤稿必须向编辑部申诉理由并征得编辑部同意ꎮ一旦稿件进入编排阶段ꎬ作者不应提出自撤稿件ꎬ在此期间因一稿两投或强行撤稿而给本刊造成不良影响和/或经济损失者ꎬ编辑部有权给以公开曝光㊁通报并实施经济赔偿ꎬ作者自行承担一切责任和后果ꎮ根据«中华人民共和国著作版权法»的相关条文ꎬ本刊编辑可对待发表的来稿按照编辑规范和专业知识进行文字加工㊁修改和删减ꎬ修改后的稿件作者须认真校对核实ꎬ修改涉及文章的核心内容时双方应进行沟通并征得作者同意ꎮ除了编辑方面的技术加工以外ꎬ作者对已经发表论文的全部内容文责自负ꎮ稿件编辑流程中编辑退回作者修改的稿件逾期2个月不修回者ꎬ视作自行撤稿ꎮ(本刊编辑部)。
Notch基因最早发现于果蝇,部分功能缺失导致翅缘缺刻(notches,图8-36)。
在胚胎发育中,当上皮组织的前体细胞中分化出神经元细胞后,其细胞表面Notch配体Delta与相邻细胞膜上的Notch结合,启动信号途径,防止其它细胞发生同样的分化,这种现象叫作侧向抑制(lateral inhibition)。
Notch突变的半合子[9]或纯合子在胚胎期死亡,其胚胎中神经组织取代了上皮组织从而使神经组织异常丰富。
Notch信号途径由Notch、Notch配体(DSL蛋白)和CSL(一类DNA结合蛋白)等组成。
Notch及其配体均为单次跨膜蛋白,当配体(如Delta)和相邻细胞的Notch结合后,Notch 被蛋白酶体切割,释放出具有核定位信号的胞质区ICN(intracellular domain of Notch),进入细胞核与CLS结合,调节基因表达。
可概括为(图8-37):Delta→Notch→酶切→ICN→进入细胞核→CLS-ICN复合体→基因转录。
图8-36 Notch缺陷引起果蝇翅缘缺刻Notch:为分子量约300KD的蛋白质,果蝇只有1个Notch基因,人类4个(Notch1-4)。
Notch的胞外区是结合配体的区域,具有不同数量的EGF样重复序列(EGF-R)和3个LNR (Lin/Notch repeats)。
胞内区由RAM(RBP-J kappa associated molecular)结构域、6个锚蛋白(cdc10/ankyrin,ANK)重复序列、2个核定位信号[10](NLS)和PEST结构域。
RAM 结构域是与CSL结合的区域,PEST结构域与Notch的降解有关。
Notch蛋白要经过三次切割,第一次在高尔基体内被furin切割为2个片断,转运到细胞膜形成异二聚体。
当配体结合到胞外区,Notch蛋白又发生两次断裂,先是被肿瘤坏死因子-α-转化酶(TNF-α-converting enzyme,TACE)切割,然后被γ-促分泌酶(γ-secretase)切割,后者需要早老蛋白(presenilin,PS)参与。