液相色谱对照品或者标准品溶液稳定性验证方案
- 格式:doc
- 大小:2.51 MB
- 文档页数:13

对照品溶液稳定性研究方案文件类型:方法研究编号:文件编号:﹡﹡页()作者分析师:审核:主管/经理批准:质量部经理:签名:日期:1.目的:本草案目的是为了研究在方法中未规定对照品溶液的效期饿稳定性。
2.背景目前,在药典中没有规定对照品溶液的效期(部分方法研究报告中提供对照品溶液的效期)。
因此,对于没有规定有效期的对照品溶液可以研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3.稳定性研究:3.1标签所有用于对照品溶液效期的溶液标签上都需要标注“用于对照品溶液效期研究”。
3.2对照品溶液配制对照品溶液的配制遵循相应的分析方法。
3.3储存条件对照品溶液密封储存在2~8℃冰箱中(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
3.4测试时间点测试时间可以用参照下表,更改的时间点需在总结报告中说明。
备注:×=测试点,【×】=可选择点3.5程序和接受标准3.5.1HPLC方法3.5.1.1对照品溶液的制备分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3.5.1.2接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显著的杂质峰,则停止对照品溶液的测试。
在每次分析前,必须首先保证系统满足方法的系统适应性(如重复性,拖尾因子,分离度,理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过2.0。
在每个分析测试点,对照品溶液的活性成分的含量与零点的差异不得过2.0。
3.5.2 UV方法……4参考文件:《中国药典》﹡﹡﹡﹡年版﹡﹡部(或其他方法号)5结果报告:对照品溶液效期研究报告结束后,需要起草研究报告总结分析数据。
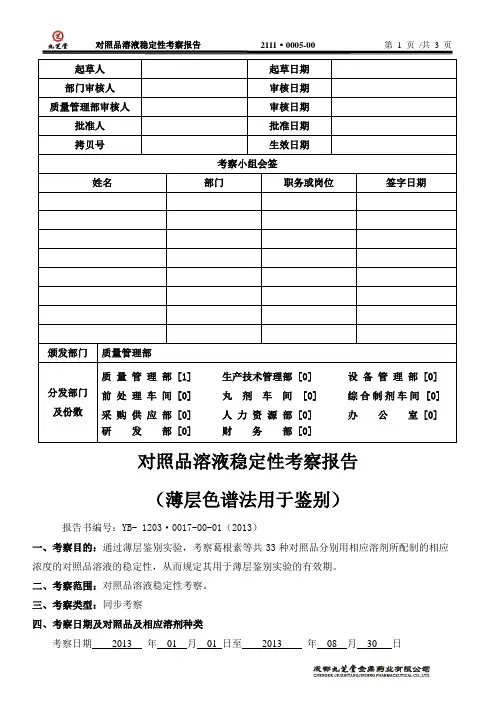
对照品溶液稳定性考察报告(薄层色谱法用于鉴别)报告书编号:YB- 1203·0017-00-01(2013)一、考察目的:通过薄层鉴别实验,考察葛根素等共33种对照品分别用相应溶剂所配制的相应浓度的对照品溶液的稳定性,从而规定其用于薄层鉴别实验的有效期。
二、考察范围:对照品溶液稳定性考察。
三、考察类型:同步考察四、考察日期及对照品及相应溶剂种类考察日期2013 年01 月01 日至2013 年08 月30 日对照品及相应溶剂种类:大黄素(甲醇液)、大黄素(乙醇液)、丹参酮IIA(乙酸乙酯液)、丹参酮IIA (甲醇液)、丹酚酸B(75%甲醇液)、丹皮酚(丙酮液)、甘草酸铵(甲醇液)、葛根素(甲醇液)、瓜氨酸(稀乙醇液)、桂皮醛(乙醇液)、华蟾酥毒配基(乙醇液)、黄芪甲苷(甲醇液)、黄芩苷(甲醇液)、黄芩素(甲醇液)、汉黄芩素(甲醇液)、苦参碱和槐定碱(混合乙醇溶液)、苦参碱(甲醇液)、氧化苦参碱(甲醇液)、苦杏仁苷(甲醇液)、芒柄花素(甲醇液)、毛蕊花糖苷(甲醇液)、没食子酸(甲醇液)、人参皂苷R0蜕皮甾酮(混合甲醇溶液)、山栀苷甲酯(乙醇液)、8-0-乙酰山栀苷甲酯(乙醇液)、芍药苷(乙醇液)、天麻素(甲醇液)、五味子甲素(三氯甲烷液)、盐酸水苏碱(无水乙醇液)、氧化苦参碱(乙醇液)、异钩藤碱(甲醇液)、栀子苷(乙醇液)、酯蟾酥毒配基(乙醇液)、梓醇(甲醇液)、盐酸小檗碱(甲醇液)、黄芩苷((甲醇液)。
五、考察过程中的偏差及漏项情况:考察试验过程中无偏差情况发生,考察项目无遗漏;考察实施过程中考察方案无修改,考察记录完整,实验结果符合考察要求,无需做进一步的补充试验。
六、考察过程总结:检验仪器设备均在检定效期内;试剂试液等实验条件均符合检验要求;人员具有相应的检验能力和资质。
整个考察过程严格按《对照品(标准品)和对照药材溶液稳定性考察操作规程》(方案编码:1203·0117-00)进行考察。

GMP文件验证篇盐酸小檗碱对照品溶液有效期验证方案方案编号: SOP-COD0400年月验证方案审批表盐酸小檗碱对照品溶液有效期验证方案目录1.目的2.背景3.稳定性研究3.1.标签3.2.对照品溶液配制3.3.贮存条件3.4.测试时间点3.5.分析方法和接受标准4.参考文件5.结果报告6.附件1.目的:确定盐酸小檗碱对照品溶液的有效期。
2.背景:因药典中没有规定对照品溶液的有效期,因此,对于没有规定有效期的盐酸小檗碱对照品溶液进行4个月的研究,来观察其稳定性,并规定其内部使用有效期。
本方案适用于常规方法—高效液相色谱法。
3. 稳定性研究:3.1.标签:用于对照品溶液效期研究的溶液瓶上需注明“用于对照品溶液效期研究”。
3.2.对照品溶液配制:根据《中国药典》2010版一部中药成方制剂—黄连上清片的含量测定项下对照品溶液制备方法。
3.3.贮存条件:按规定将配制好的盐酸小檗碱对照品溶液用封口膜封好,放在2~8℃的冰箱内贮存。
注意:用于分析前需放臵至室温。
3.4.测试时间点:分别在0天、7天、14天、30天、2个月、3个月、4个月内测试。
3.5.分析方法和接受标准:3.5.1分析方法:高效液相色谱法。
色谱条件与系统适应性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.033mol/L磷酸二氢钾溶液(35:65)为流动相;检测波长345nm,理论板数按盐酸小檗碱峰计算应不低于4000.对照品液的制备取盐酸小檗碱对照品适量,精密称定,加甲醇制成1 ml含20ug的溶液。
分别制备两份盐酸小檗碱对照品溶液(贴上“用于对照品溶液效期研究”的贴签),在零时间点,对两份对照品溶液分析两次,互相复核含量。
在零时间点以外的测试时间点,分别新鲜配制一份对照品溶液,并用新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3.5.2接受标准:在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。

xxxx药业有限公司对照品溶液稳定性研究方案起草人:日期:年月日审核人:日期:年月日批准人:日期:年月日1 目的本草案目的是为了研究在方法中未规定对照品溶液的效期的稳定性。
2 背景目前,在药典中没有规定对照品溶液的效期。
因此,对于没有规定效期的对照品溶液可以研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液效期的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3 稳定性研究:3.1 标签所有用于对照品溶液效期研究的溶液标签上都需标注“用于对照品溶液效期研究”。
3.2 对照品溶液配制对照品溶液的配制应遵循相应的产品分析方法。
3.3 储存条件对照品溶液密封储存在2~8 ℃冰箱内(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
3. 5. 1 程序方法对照品溶液的制备分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3. 5. 2接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显著的杂质峰,则停止对照品溶液的测试。
在每次分析时,必须首先保证系统满足方法的系统适用性(如重复性,拖尾因子,分离度,理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过2.0。
在每个分析测试点,对照品溶液的活性成分的含量与零点的差异不得过2.0。
4 参考文件:《药品GMP指南(质量控制实验室与物料系统)》《中国药典》2015年版一部《中国药典》2015年版二部5 结果报告:对照品溶液效期研究结束后,需要总结分析数据并起草颁布稳定性研究报告。
在对照品溶液效期研究记录复核后,报告经主管领导签字后,新的效期可以执行。

对照品溶液稳定性验证方案为了确保实验数据的准确性和可靠性,科学研究和工业生产中常常需要使用对照品来进行实验验证。
而对照品溶液的稳定性则是保证实验结果准确性的一个重要方面。
本文将介绍一种对照品溶液稳定性验证的方案,以确保实验数据的可靠性。
一、实验目的验证对照品溶液的稳定性,确保实验数据的准确性。
二、实验材料和设备1. 对照品溶液:需验证稳定性的对照品溶液;2. 贮存容器:干净、无杂质的玻璃瓶或塑料瓶;3. 取样工具:滴管、移液管等;4. 实验室常用设备:天平、离心机等。
三、实验步骤1. 准备工作在进行实验前,需对实验材料和设备进行充分的准备。
检查对照品溶液的包装是否完好无损,确保容器密封性良好。
2. 制备样品使用取样工具,从对照品溶液中取出一定量的样品。
可以根据实际需要确定取样量,确保样品的适当性和可重复性。
3. 存储条件将制备好的样品分别存储在不同的条件下,观察并比较其稳定性的差异。
常用的存储条件包括室温、低温和高温等。
根据对照品的性质和使用要求,选择合适的存储条件。
4. 存储时间对于稳定性验证实验,需要设定一定的存储时间。
一般可以选择不同的存储时间段,如24小时、48小时、72小时等。
存储时间的选取应结合实际需要和对照品的特性进行确定。
5. 实验观察在每个存储时间段结束后,观察对照品溶液的外观、颜色、浓度等性质的变化。
可以记录下来或进行拍照,以备后续分析和对比。
6. 数据分析根据观察到的实验数据,对对照品溶液的稳定性进行定性或定量的分析。
可以使用统计学方法进行数据处理,以更加客观地评估对照品溶液的稳定性。
四、实验注意事项1. 选择适当的对照品溶液,确保其代表性和稳定性。
2. 存储容器要保持干净,无杂质,防止对照品污染。
3. 存储条件和时间的选择要合理,根据实际需要进行调整。
4. 在实验过程中,应注意对照品溶液的密封性,防止外界因素的干扰。
5. 在观察实验数据时,要进行准确记录,并进行合理的数据分析。

对照品溶液稳定性考察方案文件编号起草人日期审核人日期批准人日期生效日期确认方案会签表姓名部门职务/职称备注参加验证人员姓名所在部门职务/职称验证分工验证文件题目对照品溶液稳定性考察方案编码起草人年月日审核人年月日批准人年月日目录1.简介2.验证目的3.验证依据4.验证范围5.确认小组成员与职责6.验证步骤6.1相关确认6.2验证所用仪器设备和试剂试液6.3稳定性具体步骤7.偏差分析8.验证周期9.验证结果的评审与验证结论10.文件修订与变更历史1、简介:目前,在药典中没有规定对照品溶液的有效期(部分方法报告中提供对照品溶液的有效期)。
因此,对于没有规定有效期的对照品溶液可以考察至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液的考察适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
2、验证目的:本草案目的是为了研究在方法中未规定对照品溶液的效期的稳定性。
3、验证依据:。
《中国药典》2015年版、《药品GMP指南(质量控制实验室与物料管理》4、验证范围:对照品溶液储存条件及有效期验证。
5、确认小组成员及责任姓名小组职务岗位验证职责组长质量经理负责验证管理的日常工作和验证的协调,验证方案和报告的批准。
组员QA负责监督确认实施的进展情况组员QC主管审核确认方案;督促并监督确认的正常进行;审核确认报告,对整个确认验证项目负责。
组员QC 负责确认方案的起草,负责起草验证方案和报告,按确认方案,参与确认实验的操作组员QC 按照预定方案进行确认;并做好相关记录;出现异常情况及时汇报和记录6.验证步骤6.1相关确认6.1.1所用仪器设备、玻璃器皿已经校验,且在有效期内。
6.1.2所用试剂符合《中国药典》2015年版要求,用于验证所用的对照品为同一批号最优。
6.1.3所参与验证的人员通过相应的岗位培训。
6.2验证所用仪器设备与试剂试液6.2.1仪器设备(附件1)6.2.2试剂试液(附件2)6.3稳定性考察具体步骤6.3.1标签所有用于对照品溶液效期的溶液标签上都需要标注“用于对照品溶液效期考察”。

. .对照品溶液稳定性研究方案文件类型:方法研究编号:文件编号:﹡﹡页()作者分析师:审核:主管/经理批准:质量部经理:签名:日期:1.目的:本草案目的是为了研究在方法中未规定对照品溶液的效期饿稳定性。
2.背景目前,在药典中没有规定对照品溶液的效期(部分方法研究报告中提供对照品溶液的效期)。
因此,对于没有规定有效期的对照品溶液可以研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3.稳定性研究:3.1标签所有用于对照品溶液效期的溶液标签上都需要标注“用于对照品溶液效期研教育资料word. .究”。
3.2对照品溶液配制对照品溶液的配制遵循相应的分析方法。
3.3储存条件对照品溶液密封储存在2~8℃冰箱中(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
3.4测试时间点备注:×=测试点,【×】=可选择点3.5程序和接受标准3.5.1HPLC方法3.5.1.1对照品溶液的制备分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3.5.1.2接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显著的杂质峰,则停教育资料word. .止对照品溶液的测试。
在每次分析前,必须首先保证系统满足方法的系统适应性(如重复性,拖尾因子,分离度,理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过2.0。
在每个分析测试点,对照品溶液的活性成分的含量与零点的差异不得过2.0。
3.5.2 UV方法……4参考文件:《中国药典》﹡﹡﹡﹡年版﹡﹡部(或其他方法号)5结果报告:对照品溶液效期研究报告结束后,需要起草研究报告总结分析数据。
xxxx药业有限公司对照品溶液稳定性研究方案起草人:日期:年月日审核人:日期:年月日批准人:日期:年月日1 目的本草案目的是为了研究在方法中未规定对照品溶液的效期的稳定性。
2 背景目前,在药典中没有规定对照品溶液的效期。
因此,对于没有规定效期的对照品溶液可以研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液效期的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3 稳定性研究:3.1 标签所有用于对照品溶液效期研究的溶液标签上都需标注“用于对照品溶液效期研究”。
3.2 对照品溶液配制对照品溶液的配制应遵循相应的产品分析方法。
3.3 储存条件对照品溶液密封储存在2~8 ℃冰箱内(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
3. 5. 1 程序方法对照品溶液的制备分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3. 5. 2接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显著的杂质峰,则停止对照品溶液的测试。
在每次分析时,必须首先保证系统满足方法的系统适用性(如重复性,拖尾因子,分离度,理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过2.0。
在每个分析测试点,对照品溶液的活性成分的含量与零点的差异不得过2.0。
4 参考文件:《药品GMP指南(质量控制实验室与物料系统)》《中国药典》2015年版一部《中国药典》2015年版二部5 结果报告:对照品溶液效期研究结束后,需要总结分析数据并起草颁布稳定性研究报告。
在对照品溶液效期研究记录复核后,报告经主管领导签字后,新的效期可以执行。
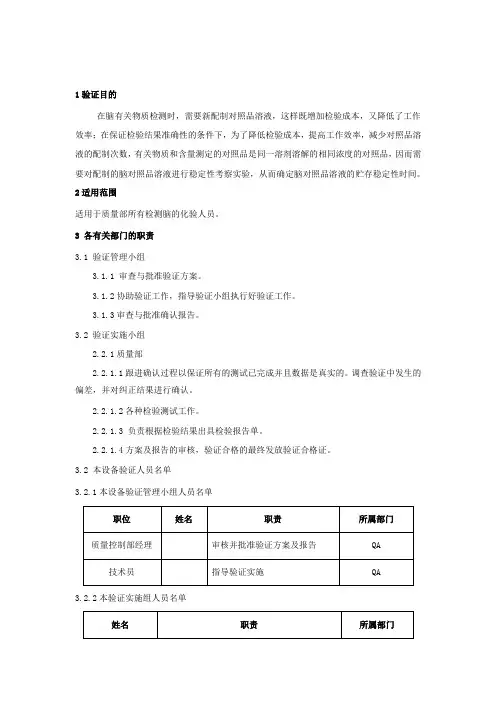
1验证目的在脑有关物质检测时,需要新配制对照品溶液,这样既增加检验成本,又降低了工作效率;在保证检验结果准确性的条件下,为了降低检验成本,提高工作效率,减少对照品溶液的配制次数,有关物质和含量测定的对照品是同一溶剂溶解的相同浓度的对照品,因而需要对配制的脑对照品溶液进行稳定性考察实验,从而确定脑对照品溶液的贮存稳定性时间。
2适用范围适用于质量部所有检测脑的化验人员。
3 各有关部门的职责3.1 验证管理小组3.1.1 审查与批准验证方案。
3.1.2协助验证工作,指导验证小组执行好验证工作。
3.1.3审查与批准确认报告。
3.2 验证实施小组2.2.1质量部2.2.1.1跟进确认过程以保证所有的测试已完成并且数据是真实的。
调查验证中发生的偏差,并对纠正结果进行确认。
2.2.1.2各种检验测试工作。
2.2.1.3 负责根据检验结果出具检验报告单。
2.2.1.4方案及报告的审核,验证合格的最终发放验证合格证。
3.2 本设备验证人员名单3.2.1本设备验证管理小组人员名单职位姓名职责所属部门质量控制部经理审核并批准验证方案及报告QA 技术员指导验证实施QA3.2.2本验证实施组人员名单姓名职责所属部门起草并实施方案,收集验证中数据并起草报告质量保证部负责设备的安装与调试并协助实施方案设备部协助验证方案的实施质量保证部3.3 试验要求3.3.1验证所用仪器设备、玻璃器皿必须经过校验,且在有效期内。
3.3.2 验证所用对照品和试剂应符合药典要求。
仪器、试剂信息表仪器名称型号编号校验日期有效期试剂名称批号级别生产厂家有效期对照品批号级别来源4、实验方法与判定4.1 对照品溶液的稳定性4.1.1 对照品溶液的配制精密量取脑对照品适量,加无水乙醇制成每1ml含脑1mg的溶液,作为对照品溶液。
4.1.2 色谱条件以交联键合聚乙二醇为固定相的毛细管柱;柱温120℃;进样口温度250℃;检测器温度250℃;分流进样,分流比10:1。

1、目的:为了确定对照品溶液的有效期特制定本办案。
2、背景:目前,在药典中没有规定对照品溶液的有效期,因此,对于没有规定有效期的对照品溶液对其进行至少三个月的稳定性考察从而确定公司内部对照品溶液的有效期。
3、稳定性研究:3.1、标签:所有用于对照品溶液有效期研究的溶液标签上是都要标注“用于对照品溶液有效期研究”。
3.2、对照品溶液的配制对照品溶液的配制遵循相应的分析方法。
3.3、储存条件对照品溶液密封储存在2-8C冰箱内,用于验证分析前需放置至室温。
3.4测试时间点:0天7天14天31天2个3月3.5、程序和接受的标准3.5.1、H PLC 方法3.5.1、对照品溶液稳定性验证程序按照相应标准中对照品配制方法分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,相互复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品和用于验证效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于验证效期的两份对照品溶液的含量值3.5.2、接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊)或者色谱图中出现显著的杂峰,则停止对照品的测试。
在每次分析时,必须首先保证系统满足方法的使用性(重现性、拖尾因子。
分离度、理论塔板数等)。
在零时间点,每份对照品溶液相互复核的结果不得过 2.0%。
在每个分析测试点对照品溶液的含量与零点的差异不得过 2.0%。
4、结论:5、附件:附录一:液相分析数据结果报告。

对照品溶液稳定性研究方案Last revision on 21 December 2020对照品溶液稳定性研究方案文件类型:方法研究编号:文件编号:﹡﹡页()作者分析师:审核:主管/经理批准:质量部经理:签名:日期:1.目的:本草案目的是为了研究在方法中未规定对照品溶液的效期饿稳定性。
2.背景目前,在药典中没有规定对照品溶液的效期(部分方法研究报告中提供对照品溶液的效期)。
因此,对于没有规定有效期的对照品溶液可以研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3.稳定性研究:标签所有用于对照品溶液效期的溶液标签上都需要标注“用于对照品溶液效期研究”。
对照品溶液配制对照品溶液的配制遵循相应的分析方法。
储存条件对照品溶液密封储存在2~8℃冰箱中(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
测试时间点测试时间可以用参照下表,更改的时间点需在总结报告中说明。
备注:×=测试点,【×】=可选择点程序和接受标准方法分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显着的杂质峰,则停止对照品溶液的测试。
在每次分析前,必须首先保证系统满足方法的系统适应性(如重复性,拖尾因子,分离度,理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过。
在每个分析测试点,对照品溶液的活性成分的含量与零点的差异不得过。
UV方法……4参考文件:《中国药典》﹡﹡﹡﹡年版﹡﹡部(或其他方法号)5结果报告:对照品溶液效期研究报告结束后,需要起草研究报告总结分析数据。
对照品溶液稳定性研究方案文件名称方案编号对照品溶液稳定性研究方案对照品溶液稳定性研究方案1.目的:本草案目的是为了研究在HPLC含量测定特定存放条件下方法中未规定对照品溶液的有效期稳定性。
2.背景:目前,在2010版《中国药典》中没有规定对照品溶液的有效期。
对照品因量小价格昂贵,为获取企业利润最大化,避免不必要的费用支出。
因此,对于没有规定有效期的对照品溶液研究至少一个月的时间,来确定对照品溶液的稳定性。
对于对照品溶液的研究适用于常规分析方法,例如液相,气相,薄层,紫外等分析方法。
3.稳定性研究:3.1标签所有用于对照品溶液效期的溶液标签上都需要标注“用于对照品溶液效期研究”。
3.2对照品溶液配制对照品溶液的配制遵循相应的分析方法。
3.3储存条件对照品溶液密封储存在2,8?冰箱中(其他储存条件需在总结报告中说明),用于验证分析前需放置至室温。
3.4测试时间点测试时间可以用参照下表,更改的时间点需在总结报告中说明。
测试时间点0天 7天 14天 31天 2个月 3个月× × × × 【×】【×】备注:×=测试点,【×】=可选择点3.5程序和接受标准3.5.1HPLC方法3.5.1.1对照品溶液的制备分别制备两份对照品溶液,在零时间点,对每份对照品溶液分析两次,互相复核。
在零时间点以外的测试时间点,新鲜配制一份对照品溶液。
对新鲜配制的对照品溶液和用于研究1文件名称方案编号对照品溶液稳定性研究方案效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3.5.1.2接受标准在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如果溶液异常(如出现浑浊),或者色谱图中出现显著的杂质峰,则停止对照品溶液的测试。
在每次分析前,必须首先保证系统满足方法的系统适应性(如重复性,拖尾因子,分离度,理论塔板数等等)。
对照品溶液稳定性试验规程
1.引言
制定本规程的目的是对产品检验时配制的部分对照品溶液进行稳定性考察,以确定对照品溶液的使用时间,保证药品检验的准确。
2.依据
国家药品监督管理局«药品生产质量管理规范»(1998年修订)第七十五条和«中国药典»二部附录XIXA
3.适用范围
本规程适用于本公司产品质量标准中配制的对照品溶液。
4.责任
QC经理,检验员对本规程实施负责。
5.程序
5.1考察用对照品溶液的配制应按检验标准中规定的方法配制。
配制成的对照品溶液贮存在容量瓶中。
5.2考察的对照品为正在使用的批号。
更换新批号的对照品使用时应重新做稳定性实验。
5.3测定次数设为1天,3天,6天,10天。
5.4测试条件:室温25±2℃,相对湿度40%-70%
5.5测试项目:性状,含量。
5.6数据评估:每次含量测定2次,每次的平均值和第1次的平均
值比较,相对偏差不得过2%。
5.7结果评估:测量误差在规定范围内,则可以使用到相应的天数,
否则不能适用。
GMP文件验证篇
盐酸小檗碱对照品溶液有效期验证方案
方案编号: SOP-COD0400
年月
验证方案审批表
验证方案名称盐酸小檗碱对照品溶液有效期验证方案
验证方案编号SOP—COD0400
方案起草人起草日期
方案审核部门审核人审核日期审核意见化验室
质量部
批准意见
批准人
批准日期
执行日期
盐酸小檗碱对照品溶液有效期验证方案目录
1.目的
2.背景
3.稳定性研究
.标签
.对照品溶液配制
.贮存条件
.测试时间点
.分析方法和接受标准
4.参考文件
5.结果报告
6.附件
1.目的:确定盐酸小檗碱对照品溶液的有效期。
2.背景:因药典中没有规定对照品溶液的有效期,因此,对于没有规定有效期的盐酸小檗碱对照品溶液进行4个月的研究,来观察其稳定性,并规定其内部使用有效期。
本方案适用于常规方法—高效液相色谱法。
3. 稳定性研究:
.标签:用于对照品溶液效期研究的溶液瓶上需注明“用于对照品溶液效期研究”。
.对照品溶液配制:根据《中国药典》2010版一部中药成方制剂—黄连上清片的含量测定项下对照品溶液制备方法。
.贮存条件:按规定将配制好的盐酸小檗碱对照品溶液用封口膜封好,放在2~8℃的冰箱内贮存。
注意:用于分析前需放置至室温。
.测试时间点:分别在0天、7天、14天、30天、2个月、3个月、4个月内测试。
.分析方法和接受标准:
3.5.1分析方法:高效液相色谱法。
色谱条件与系统适应性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈L磷酸二氢钾溶液(35:65)为流动相;检测波长345nm,理论板数按盐酸小檗碱峰计算应不低于4000.
对照品液的制备取盐酸小檗碱对照品适量,精密称定,加甲醇制成1 ml含20ug的溶液。
分别制备两份盐酸小檗碱对照品溶液(贴上“用于对照品溶液效期研究”的贴签),在零时间点,对两份对照品溶液分析两次,互相复核含量。
在零时间点以外的测试时间点,分别新鲜配制一份对照品溶液,并用新鲜配制的对照品溶液和用于研究效期的两份对照品溶液分析两次。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
3.5.2接受标准:在每次分析前,观察测试用的对照品溶液与新鲜配制的对照品溶液的外观是否一致。
如出现浑浊,或者色谱图中出现显著的杂质峰,则停止对照品溶液的测试。
在每次分析时,必须首先保证系统满足该方法的系统适用性(重复性、拖尾因子、分离度、理论塔板数等等)。
在零时间点,每份对照品溶液互相复核的结果不得过%,在每个分析测试点,对照品溶液的活性成分含量与零点的差异不得过%。
4.参考文件:《中国药典》2010版一部
5.验证报告
6.附件:液相分析数据结果报告
液相分析数据结果报告
分析方法:分析项目:
时间点
重量
(mg)
峰面积
含量
(%)
含量平均
值(%)
与零时间点的差
(%)
零点
()天
新配制的对照品溶液1 编号:()天
新配制的对照品溶液2 编号:()天
新配制的对照品溶液2 编号:()天
新配制的对照品溶液3 编号:()天
新配制的对照品溶液4 编号:()天
百度文库 - 让每个人平等地提升自我
新配制的对照品溶液4
编号:
复核者: 日期:
盐酸小檗碱对照品溶液稳定性研究报告
醉鱼草皂苷Ⅵb 对照品溶液有效期验证方案
方案编号: SOP-COD0100
年 月
年 月
盐酸小檗碱对照品溶液稳定性研究报告
文件类型盐酸小檗碱对照品溶液有效期方法研究报告测试方法测试项目
报告审核部门审核人审核日期审核意见化验室
质量部
报告批准者批准日期批准意见执行日期
盐酸小檗碱对照品溶液稳定性研究报告目录
1.目的
2.设备
3.对照品名称和测试过程
4.贮存条件
5.结果
6.变更
7.结论
1.目的:依据盐酸小檗碱对照品溶液稳定性研究方案(SOP—COD0400),评估黄连上清片中含量测定方法中对照品溶液的稳定性,制订盐酸小檗碱对照品溶液的内部使用有效期。
设备名称型号
冰箱
HPLC
天平
3.对照品名称和测试过程:
时间点对照品溶液编号
对照品
名称批号重量(mg)
0天对照品溶液Ⅰ盐酸小檗碱对照品溶液Ⅱ盐酸小檗碱
7天新配制的对照品溶液1 盐酸小檗碱
14天新配制的对照品溶液2盐酸小檗碱
30天新配制的对照品溶液3盐酸小檗碱
2个月新配制的对照品溶液4盐酸小檗碱
3个月新配制的对照品溶液5盐酸小檗碱
测试过程:高效液相色谱法。
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈L磷酸二氢钾溶液(35:65)为流动相;检测波长345nm,理论板数按盐酸小檗碱峰计算应不低于4000.
对照品液的制备取盐酸小檗碱对照品适量,精密称定,加甲醇制成1 ml含20ug的溶液。
测定法分别精密吸取盐酸小檗碱对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定。
4.贮存条件:验证用的盐酸小檗碱对照品溶液用封口膜封好,放在2~8℃的冰箱内贮存。
5.结果:在零时间点,对两份对照品溶液分析两次,互相复核含量。
在零时间点以外的测试时间点。
用新鲜配制的对照品溶液的平均响应值,来重新计算用于研究效期的两份对照品溶液的含量值。
结果见附件1。
具体计算见附页。
6.结论:
附件1 液相分析数据结果报告
分析方法:分析项目:
时间点
重量
(mg)
峰面积
含量
(%)
含量平均
值(%)
与零时间点的差
(%)
零点
()天
新配制的对照品溶液1 编号:()天
新配制的对照品溶液2 编号:()天
新配制的对照品溶液2 编号:()天
新配制的对照品溶液3 编号:()天
新配制的对照品溶液4 编号:()天
新配制的对照品溶液4 编号:
复核者:日期:。