对甲苯乙酮的制备
- 格式:docx
- 大小:2.06 MB
- 文档页数:10

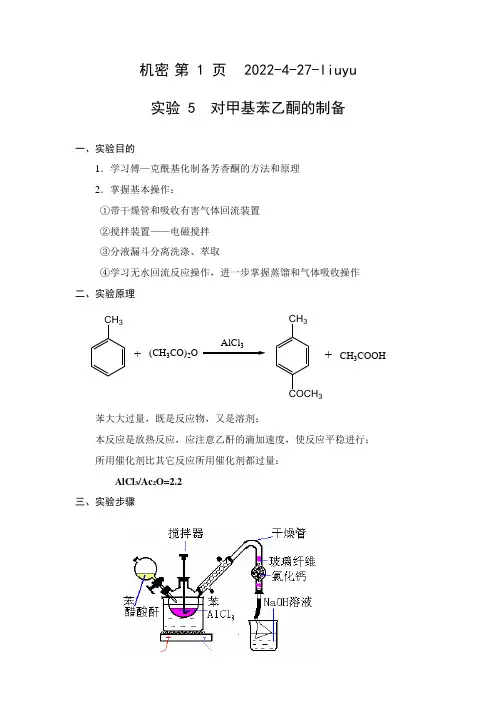
机密 第 1 页 2022-4-27-liuyu实验 5 对甲基苯乙酮的制备一、实验目的1.学习傅—克酰基化制备芳香酮的方法和原理2.掌握基本操作:①带干燥管和吸收有害气体回流装置②搅拌装置——电磁搅拌③分液漏斗分离洗涤、萃取④学习无水回流反应操作,进一步掌握蒸馏和气体吸收操作二、实验原理苯大大过量,既是反应物,又是溶剂;本反应是放热反应,应注意乙酐的滴加速度,使反应平稳进行; 所用催化剂比其它反应所用催化剂都过量:AlCl 3/Ac 2O=2.2三、实验步骤CH 3CH 3(CH 3CO)2O AlCl 33CH 3COOH++2、实验步骤在分别装有恒压滴液漏斗,球形冷凝管的100 mL的三颈瓶中(冷凝管上端装一氯化钙干燥管,干燥管再与氯化氢气体吸收装置相连)。
迅速称取10 g 经研细的无水三氯化铝加入三颈瓶中,再加入25mL 无水甲苯,在搅拌下自滴液漏斗慢慢滴加3mL 的新蒸乙酸酐和3 mL 无水甲苯的混合液(先加几滴,待反应发生后再继续滴加),控制滴加速度勿使反应过于激烈,以三颈瓶稍热为宜(60 ℃)。
边滴加边搅拌,约10-15min 滴加完毕。
加完后,在沸水浴上(90-95 ℃)回流15-20min,直至不再有氯化氢气体逸出为止。
将反应物冷至室温,在搅拌下将反应物倒入盛有15mL 浓盐酸和20g 碎冰的烧杯中进行分解(在通风橱中进行)。
当固体完全溶解后,将混合物转入分液漏斗,分出有机层,水层每次用20 mL 甲苯萃取两次(10-1)。
合并有机层和甲苯萃取液,依次用等体积(10 mL)的10%氢氧化钠溶液和(10 mL)水洗涤一次,用无水硫酸镁干燥。
将干燥后的粗产物先在水浴上蒸去甲苯,当温度上升至140℃左右时,停止加热,减压蒸馏,收集〖?〗℃馏分,产量约?g。
有机层粗产物水层苯萃取2次(4ml/次)合并苯层水层碱洗水洗水层有机层(弃去)无水硫酸镁干燥澄清有机层蒸馏回收溶剂苯改用空气冷凝管蒸馏收集195~202℃馏分称量(无色透明油状液体)四、实验注意事项:①装置仪器和试剂应无水,否则反应失败;②滴加乙酐时应控制速度,使反应平稳进行;③注意反应终点和反应混合物处理时一定在通风橱内进行。


有机实验思考题解析实验一熔点的测定(P56-60)思考题P.60 (1),(2),(3)题。
(1)如何验证两种熔点相近的物质是否为同一种物质?答:把它们混合,测该混合物的熔点,若熔点仍不变,才能认为它们为同一物质。
若混合物熔点降低,熔程增大,则说明它们属于不同的物质。
(2)熔点毛细管是否可以重复使用?答:不可以。
(3)测溶点时,如有下列情况将产生什么结果?①溶点管太厚②溶点管不干净③样品未完全干燥或含有杂质④样品研得不细⑤样品装得不紧密⑥加热太快⑦样品装得太多⑧读数太慢答:①溶点管太厚,热传导时间长,会产生熔点偏高。
②溶点管不干净,能产生4-10℃的误差。
③样品未完全干燥或含有杂质,会使熔点偏低,熔程变大。
④样品研得不细,会产生空隙,不易传热,造成熔程变大。
⑤样品装得不紧密,会产生空隙,不易传热,造成熔程变大。
⑥加热太快,熔点偏高。
⑦样品装得太多,会造成熔程变大,熔点偏高。
⑧读数太慢,熔点偏高。
实验二蒸馏和沸点的测定(P78-81)思考题P.81(1),(2),(3)题。
(1)蒸馏时温度计的位置偏高和偏低,馏出液的速度太慢或太快,对沸点的读数有何影响?答:沸点的读数会偏高或偏低。
(2)如果馏出液的物质易受潮分解、易挥发、易燃、易爆或有毒,应该采取什么办法?答:如果馏出液的物质易挥发、易燃、易爆或有毒,必须选择无明火操作的热浴;尾气通入下水道,收集瓶放在水浴或冰水浴中。
如果馏出液的物质易受潮分解,可在收集瓶中预先加入干燥剂。
(3)蒸馏时为什么要加沸石,如果加热后才发现未加入沸石,应该怎样处理?答:蒸馏过程中上升的气泡增大得非常快,甚至将液体冲出瓶外,这种不正常的沸腾称为“暴沸”。
为了防止在蒸馏过程中的“暴沸”现象,常加入沸石,或一端封口的毛细管,以引入汽化中心,产生平稳沸腾。
当加热后发现未加入沸石时,千万不能匆忙加入沸石,会引起猛烈的暴沸,液体易冲出瓶口。
应该移去热源,使液体冷却至沸点以下后才能加入。
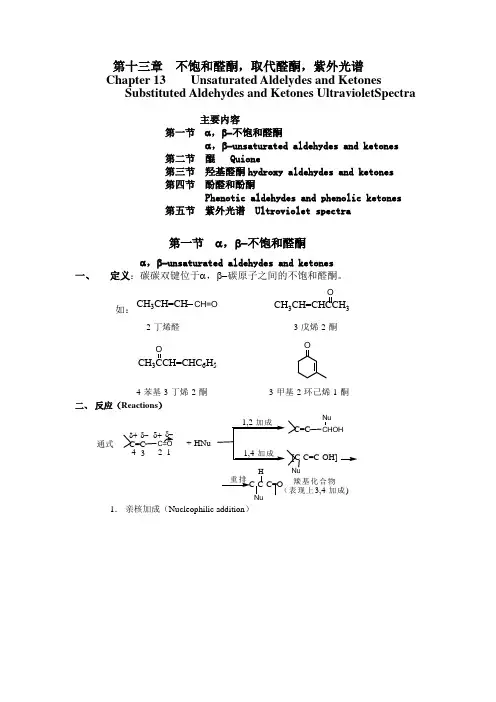
第十三章 不饱和醛酮,取代醛酮,紫外光谱 Chapter 13 Unsaturated Aldelydes and KetonesSubstituted Aldehydes and Ketones UltravioletSpectra主要内容第一节 α,β-不饱和醛酮α,β-unsaturated aldehydes and ketones 第二节 醌 Quione第三节 羟基醛酮hydroxy aldehydes and ketones 第四节 酚醛和酚酮Phenotic aldehydes and phenolic ketones第五节 紫外光谱 Ultroviolet spectra第一节 α,β-不饱和醛酮α,β-unsaturated aldehydes and ketones 一、 定义:碳碳双键位于α,β-碳原子之间的不饱和醛酮。
如:CH 3CH=CH CH=OOCH 3CH=CHCCH 32-丁烯醛 3-戊烯-2-酮 CH 36H 5OO4-苯基-3-丁烯-2-酮 3-甲基-2-环己烯-1-酮二、 反应(Reactions )通式C=C C=O4321δ+δ-δ-δ++HNuC=CCHOHNu[C-C=C-OH]Nu重排C-C-C=ONuH 羰基化合物(表现上3,4-加成)1. 亲核加成(Nucleophilic addition )C=C-C=O(1,2-加成产物为主)甲基酮(1,2-加成为主)其他酮(1,4-加成为主)例:C 6H 5CH=CHCOC 6H 5KCN ,HOAcEtOHC 6H 5CH(CN)CH 2COC 6H 5(93-96%)O+ HCNCN O(85%)C 6H 5CH=CHCOC 6H 51)C6H 5Li 2)H 2OC 6H 5CH=CHC(OH)(C 6H 5)2(75%)O+(CH 3)2CuLi22O(98%)2. 亲电加成(electrophilic addition )CH 3δ+4CH 3CH(Cl)CH 2COCH 3CH 3CH(Br)CH(Br)COCH 3反应速率比单烯烃及共轭二烯烃慢。

杀菌剂氰霜唑的合成进展两'l『召循壤PesticideScienceandAdministration2009,30(10)许诚,丁秀丽,李宗英,鲁鸣久(西安近代化学研究所,西安710065) AdvancesintheSynthesisofCyazofamidXuCheng,DingXiuli,LIZongying,LuMin~iu(Xi'anModernChemistryResearchInstitute,Xi'an710065,China)Abstract:CyazofamidisanovelfungicidewhichisusedtocontroltheOomycetediseases.Aft ersummarizingandcomparingthesyntheticroutesofcyazofamidinthispaper,aroutesuitablef orindustrializationwasselected.Keywords:fungicide;cyazofamid;synthesis摘要:本文综述了氰霜唑的主要合成路线,并对各条路线作出了评述,从而选出一条适合工业化的路线.关键词:杀菌剂;氰霜唑;合成中图分类号:TQ450.6;$482.1 文献标识码:A 文献编号:1002-5480(2009)10-40-02氰霜唑…是日本石原株式会社研制与BASF共同开发的新一代咪唑类杀菌剂.化学名为 4 一氯一 2 一氰基一N.N —二甲基一 5 一P 一甲苯基咪唑一l 一磺酰胺.化学结构式:/c"3wn.cl/〜N氰霜唑[Z]作用机理是作用于线粒体电子传递络合物川Qi部位的第1个农用杀菌剂,用于防治以霜霉病,疫病为代表的卵菌纲病害,具有很高的杀菌活性.对疫霉菌生活史的各个成长阶段包括孢子囊的形成,萌发,卯孢子的形成, 游动孢子的释放,移动以及菌丝的生长等都具有很高的抑制作用.氰霜唑使用剂量低,对人畜低毒.目前.国外对氰霜唑的研究活跃,国内对其研究报道不多.本文综述了氰霜唑的合成方法.并对各种方法进行了比较分析.总结出各自的优缺点.1合成路线氰霜唑的合成根据起始原料和重要中间体合成途径的不同.主要有4种合成方法.1.1以对甲苯乙酮为原料的丁基锂路线[以对甲苯乙酮为原料.经卤化和甲酰得到4 一甲基咪唑.再将化合物在一70C下,滴加正丁--二 a 一.=l_fHBr收稿日期:2009—04—2340琴Ij---H~cn 五I.l.图 1 丁基锂路线合成氰霜唑;511『彳霍理PesticideScienceandAdministration2009,30(zo) 基锂,反应15h得到化合物,随后与羟胺溶于吡啶中,室温滴加乙酐,IO0~C反应12h,得到2一氰基一 5 一(4 一甲基苯基)咪唑,最后氯化和磺酰胺化得到氰霜唑.1.2以对甲苯乙酮为原料的乙二醛路线[33c「CH=B』图2乙二醛路线合成氰霜唑以对甲苯乙酮为原料,经过卤化得到2,2一卤代一4'一甲基苯乙酮,再与乙二醛和羟胺在甲醇中反应24h,得到1 一羟基一 4 一(4 一甲基苯基)2 一甲肟基咪唑一3 一氧化物,然后脱水与氯化得到 2 一氰基一4 一氯一5 一(4 一甲基苯基)咪唑,最后磺酰胺化得到氰霜唑.1.3以OL 一氨基酮为原料的酰亚胺路线[]NH,.H)yddine2)(CHsc0)20ClSO2N(CH3)2K'C0tC'2H50)2CHCtOCH3)~N cH2NH2HCINaOCH3图3酰亚胺路线合成氰霜唑将二乙氧乙酰亚胺于室温滴加至Or,—氨基酮的含NaOCH,的甲醇溶液中,室温反应1h 然后回流2h.中和环化得到2 一乙氧甲基一 4 一对甲苯咪唑.随后肟化,脱水得到 2 一氰基一5一(4一甲基苯基)咪唑,经氯化,二甲基磺酰胺化得到氰霜唑.1-4 以一氨基酮为原料的乙二氰路线[以or.—氨基酮为原料.与乙二氰的DMF溶液H,/JI,I .H_』—L—OH图 4 乙二氰路线合成氰霜唑在吡啶中回流3h,环化生成2 一氰基一 5 一(4 一甲基苯基)咪唑,再氯化与磺酰胺化生成氰霜唑.2 合成方法的评述2.1 以对甲苯乙酮为原料的丁基锂路线原料来源丰富,易得,但反应路线长,单元反应多, 所用试剂丁基锂很活泼,反应温度低,反应条件要求苛刻.对设备的要求高,不适宜工业化生产.2.2以对甲苯乙酮为原料的乙二醛路线原料来源丰富,易得,成本低,反应条件温和,不需要特殊设备,虽然反应路线较长,但原子经济性好,总收率较高,有好的工业化前景.2.3以0【一氨基酮为原料的酰亚胺路线路线较长.工艺条件较简单,但所需试剂酰亚胺无工业化产品.并且制备闲难.造成工业生产成本上升.2.4以OL 一氨基酮为原料的乙二氰路线虽然线路短,单元反应少,反应条件温和,但是原料之一为乙二氰.该物质为剧毒气体,操作难度大.对工业化生产很不利.3结论可见,在文献报道的氰霜唑的合成路线中, 比较适合工业化生产的路线为乙二醛路线是以对甲苯乙酮为原料.经卤化得到Ot 一卤代对甲苯乙酮.再与羟胺和乙二醛反应,然后经脱水, 氯化,磺酰胺化要得到氰霜唑.参考文献1 刘长令主编.世界农药大全一杀菌剂卷[M]. 北京:化学工业出版社.2006.2Nasa,Komy~i,Suzukieta1.Imidazolecompoundsandbio—cidalcompositionscomprisingthesame下转第29 页)4l 一N帐蔼钟彳,} 霍理PesticideScienceandAdministration2009,30(10)1.4 建立举报制度.公开举报电话向社会公开举报电话.对经营不规范,不合格标签农药产品的违法行为,要向社会曝光,追根寻源, 依法予以处理,凡是抽检不合格的经营单位要责令限期整改.农药管理部门在切实履行自身职责的同时,充分发挥辖区内技术监督,工商行政管理,公安等职能部门的作用,及时将抽检结果向他们通报,争取各方面的支持,共同规范市场.确保全市农药市场标签抽检合格率保持在89%以上2 标签抽查的做法2.1突击重点,随机抽取我市现有耕地面积460 万亩.农药经营单位795 家根据作物主栽品种和农民用药习惯,每年采取定期抽查和不定期抽查相结合的形式.结合不同作物的生产季节,重点抽查当地使用量大的除草剂,杀菌剂,杀虫剂品种.突出对疑似含有高毒成分产品的抽查.为确保抽查产品的真实性和合法性抽查方法严格按照相关法定程序.到农药经营单位随机抽取.2.2突出抓好农药标签四查一查农药"三证"号:"i证"指农药登记证号,产品标准证号,生产批准证号.国产农药必须"i 证" 齐全,原装进口农药直接销售的可以不标农药生产许可证号或农药生产批准文件号,产品标准证号.二查使用范同:标签上标注的适用作物和防治对象必须一致.防治对象与施用作物不复的,随意扩大防治范围和防治对象的都属于不允许销售范嗣.三查标签内容:包括农药名称,有效成分名称,含量和剂型.从2008 年7月1日起生产的农药.只使用农药通用名称取消商品名称.看标注的有效成分名称,含量及剂型是否清晰:看使用技术和方法,毒性标识,注意事项,中毒急救措施,贮存和运输方法,农药类别,象形网等内容是否齐全.四查产品合格证:农药产品的包装箱内,都应附有产品出厂检验合格证.检查时一并进行查看. 3取得的成效3.1农药标签合格率逐年提高2006年一2008 年全市共抽查农药标签466 个,统计分析结果表明2006 年全市抽查标签150 个,合格标签119 个,合格率为79.3%:2007年全市抽查标签134个,合格标签1l5 个,合格率为85.82%; 2008年全市抽查标签182个.合格标签163 个, 合格率为89.56%.全市标签合格率平均每年提高 4 个百分点以上3.2农产品质量明显提高由于狠抓农药标签抽查判定工作,确保了全市农产品质量安全, 据承德市农产品检测中心统计:2006 年一2008 年全市开展农产品检测个3l3 批次,3731 个样品其中2006年抽检92批次,1123个样品,检测合格率98.42%;2007年抽检96批次, 1252个样品,检测合格率97.44%:2008年抽检125批次,1356个样品,检测合格率99-3%, 使全市的蔬菜检测合格率连续三年居全省前i 名,农产品质量安全水平明显增强.+"+一+一+-+一+-+-+-+-+-+-+-+-+ 一+-+-+ 一+一+一+一+一+一+一+一+一+一+ +一+-+-+-+-+-+-+ 一+一+-+一+一+一+一+一+(上接第41 页)[P].EP0298196A1,1989—11-0l 3Jonishi,Hisayoshi,Kanamorleta1.Processforproducing 1-substituted — 2 一cyanoimidazolecompounds[PJ.EP070- 5823A1.1995-09〜06.4TakeshiOHSHIMA,TerumasaKOMYOJIA,ShigeruMIT-ANI.Developmentofanovelfungicide,cyazofamid[J].J.Pestic.Sci,2004,29:136~138.5Fujili,Yasuhiro,Tonimuraeta1.Processforproducing2- cyanoimidazolecompounds[P].EP0653421A1,1994—11—03.29—。
杀菌剂氰霜唑的合成进展两'l『召循壤PesticideScienceandAdministration2009,30(10)许诚,丁秀丽,李宗英,鲁鸣久(西安近代化学研究所,西安710065) AdvancesintheSynthesisofCyazofamidXuCheng,DingXiuli,LIZongying,LuMin~iu(Xi'anModernChemistryResearchInstitute,Xi'an710065,China)Abstract:CyazofamidisanovelfungicidewhichisusedtocontroltheOomycetediseases.Aft ersummarizingandcomparingthesyntheticroutesofcyazofamidinthispaper,aroutesuitablef orindustrializationwasselected.Keywords:fungicide;cyazofamid;synthesis摘要:本文综述了氰霜唑的主要合成路线,并对各条路线作出了评述,从而选出一条适合工业化的路线.关键词:杀菌剂;氰霜唑;合成中图分类号:TQ450.6;$482.1 文献标识码:A 文献编号:1002-5480(2009)10-40-02氰霜唑…是日本石原株式会社研制与BASF共同开发的新一代咪唑类杀菌剂.化学名为 4 一氯一 2 一氰基一N.N —二甲基一 5 一P 一甲苯基咪唑一l 一磺酰胺.化学结构式:/c"3wn.cl/〜N氰霜唑[Z]作用机理是作用于线粒体电子传递络合物川Qi部位的第1个农用杀菌剂,用于防治以霜霉病,疫病为代表的卵菌纲病害,具有很高的杀菌活性.对疫霉菌生活史的各个成长阶段包括孢子囊的形成,萌发,卯孢子的形成, 游动孢子的释放,移动以及菌丝的生长等都具有很高的抑制作用.氰霜唑使用剂量低,对人畜低毒.目前.国外对氰霜唑的研究活跃,国内对其研究报道不多.本文综述了氰霜唑的合成方法.并对各种方法进行了比较分析.总结出各自的优缺点.1合成路线氰霜唑的合成根据起始原料和重要中间体合成途径的不同.主要有4种合成方法.1.1以对甲苯乙酮为原料的丁基锂路线[以对甲苯乙酮为原料.经卤化和甲酰得到4 一甲基咪唑.再将化合物在一70C下,滴加正丁--二 a 一.=l_fHBr收稿日期:2009—04—2340琴Ij---H~cn 五I.l.图 1 丁基锂路线合成氰霜唑;511『彳霍理PesticideScienceandAdministration2009,30(zo) 基锂,反应15h得到化合物,随后与羟胺溶于吡啶中,室温滴加乙酐,IO0~C反应12h,得到2一氰基一 5 一(4 一甲基苯基)咪唑,最后氯化和磺酰胺化得到氰霜唑.1.2以对甲苯乙酮为原料的乙二醛路线[33c「CH=B』图2乙二醛路线合成氰霜唑以对甲苯乙酮为原料,经过卤化得到2,2一卤代一4'一甲基苯乙酮,再与乙二醛和羟胺在甲醇中反应24h,得到1 一羟基一 4 一(4 一甲基苯基)2 一甲肟基咪唑一3 一氧化物,然后脱水与氯化得到 2 一氰基一4 一氯一5 一(4 一甲基苯基)咪唑,最后磺酰胺化得到氰霜唑.1.3以OL 一氨基酮为原料的酰亚胺路线[]NH,.H)yddine2)(CHsc0)20ClSO2N(CH3)2K'C0tC'2H50)2CHCtOCH3)~N cH2NH2HCINaOCH3图3酰亚胺路线合成氰霜唑将二乙氧乙酰亚胺于室温滴加至Or,—氨基酮的含NaOCH,的甲醇溶液中,室温反应1h 然后回流2h.中和环化得到2 一乙氧甲基一 4 一对甲苯咪唑.随后肟化,脱水得到 2 一氰基一5一(4一甲基苯基)咪唑,经氯化,二甲基磺酰胺化得到氰霜唑.1-4 以一氨基酮为原料的乙二氰路线[以or.—氨基酮为原料.与乙二氰的DMF溶液H,/JI,I .H_』—L—OH图 4 乙二氰路线合成氰霜唑在吡啶中回流3h,环化生成2 一氰基一 5 一(4 一甲基苯基)咪唑,再氯化与磺酰胺化生成氰霜唑.2 合成方法的评述2.1 以对甲苯乙酮为原料的丁基锂路线原料来源丰富,易得,但反应路线长,单元反应多, 所用试剂丁基锂很活泼,反应温度低,反应条件要求苛刻.对设备的要求高,不适宜工业化生产.2.2以对甲苯乙酮为原料的乙二醛路线原料来源丰富,易得,成本低,反应条件温和,不需要特殊设备,虽然反应路线较长,但原子经济性好,总收率较高,有好的工业化前景.2.3以0【一氨基酮为原料的酰亚胺路线路线较长.工艺条件较简单,但所需试剂酰亚胺无工业化产品.并且制备闲难.造成工业生产成本上升.2.4以OL 一氨基酮为原料的乙二氰路线虽然线路短,单元反应少,反应条件温和,但是原料之一为乙二氰.该物质为剧毒气体,操作难度大.对工业化生产很不利.3结论可见,在文献报道的氰霜唑的合成路线中, 比较适合工业化生产的路线为乙二醛路线是以对甲苯乙酮为原料.经卤化得到Ot 一卤代对甲苯乙酮.再与羟胺和乙二醛反应,然后经脱水, 氯化,磺酰胺化要得到氰霜唑.参考文献1 刘长令主编.世界农药大全一杀菌剂卷[M]. 北京:化学工业出版社.2006.2Nasa,Komy~i,Suzukieta1.Imidazolecompoundsandbio—cidalcompositionscomprisingthesame下转第29 页)4l 一N帐蔼钟彳,} 霍理PesticideScienceandAdministration2009,30(10)1.4 建立举报制度.公开举报电话向社会公开举报电话.对经营不规范,不合格标签农药产品的违法行为,要向社会曝光,追根寻源, 依法予以处理,凡是抽检不合格的经营单位要责令限期整改.农药管理部门在切实履行自身职责的同时,充分发挥辖区内技术监督,工商行政管理,公安等职能部门的作用,及时将抽检结果向他们通报,争取各方面的支持,共同规范市场.确保全市农药市场标签抽检合格率保持在89%以上2 标签抽查的做法2.1突击重点,随机抽取我市现有耕地面积460 万亩.农药经营单位795 家根据作物主栽品种和农民用药习惯,每年采取定期抽查和不定期抽查相结合的形式.结合不同作物的生产季节,重点抽查当地使用量大的除草剂,杀菌剂,杀虫剂品种.突出对疑似含有高毒成分产品的抽查.为确保抽查产品的真实性和合法性抽查方法严格按照相关法定程序.到农药经营单位随机抽取.2.2突出抓好农药标签四查一查农药"三证"号:"i证"指农药登记证号,产品标准证号,生产批准证号.国产农药必须"i 证" 齐全,原装进口农药直接销售的可以不标农药生产许可证号或农药生产批准文件号,产品标准证号.二查使用范同:标签上标注的适用作物和防治对象必须一致.防治对象与施用作物不复的,随意扩大防治范围和防治对象的都属于不允许销售范嗣.三查标签内容:包括农药名称,有效成分名称,含量和剂型.从2008 年7月1日起生产的农药.只使用农药通用名称取消商品名称.看标注的有效成分名称,含量及剂型是否清晰:看使用技术和方法,毒性标识,注意事项,中毒急救措施,贮存和运输方法,农药类别,象形网等内容是否齐全.四查产品合格证:农药产品的包装箱内,都应附有产品出厂检验合格证.检查时一并进行查看. 3取得的成效3.1农药标签合格率逐年提高2006年一2008 年全市共抽查农药标签466 个,统计分析结果表明2006 年全市抽查标签150 个,合格标签119 个,合格率为79.3%:2007年全市抽查标签134个,合格标签1l5 个,合格率为85.82%; 2008年全市抽查标签182个.合格标签163 个, 合格率为89.56%.全市标签合格率平均每年提高 4 个百分点以上3.2农产品质量明显提高由于狠抓农药标签抽查判定工作,确保了全市农产品质量安全, 据承德市农产品检测中心统计:2006 年一2008 年全市开展农产品检测个3l3 批次,3731 个样品其中2006年抽检92批次,1123个样品,检测合格率98.42%;2007年抽检96批次, 1252个样品,检测合格率97.44%:2008年抽检125批次,1356个样品,检测合格率99-3%, 使全市的蔬菜检测合格率连续三年居全省前i 名,农产品质量安全水平明显增强.+"+一+一+-+一+-+-+-+-+-+-+-+-+ 一+-+-+ 一+一+一+一+一+一+一+一+一+一+ +一+-+-+-+-+-+-+ 一+一+-+一+一+一+一+一+(上接第41 页)[P].EP0298196A1,1989—11-0l 3Jonishi,Hisayoshi,Kanamorleta1.Processforproducing 1-substituted — 2 一cyanoimidazolecompounds[PJ.EP070- 5823A1.1995-09〜06.4TakeshiOHSHIMA,TerumasaKOMYOJIA,ShigeruMIT-ANI.Developmentofanovelfungicide,cyazofamid[J].J.Pestic.Sci,2004,29:136~138.5Fujili,Yasuhiro,Tonimuraeta1.Processforproducing2- cyanoimidazolecompounds[P].EP0653421A1,1994—11—03.29—。

实验八1一对甲苯基一3一苯基一2一丙烯一1一酮(1一p—methyphenyl一3一phenyl—2一propene一1一one) 由苯甲醛与对甲苯乙酮经Claisen-Schmidt反应制备1一对甲苯基一3一苯基一2一丙烯一1一酮,事实表明,在一对顺反异构体中,E型产物为主要产物。
【反应式】【试剂】对甲苯乙酮3.4 g(3.4 mL,0.025 mol),苯甲醛2.7 g(2.6 mL,0.025 mol),氢氧化钠1.3 g,95%乙醇溶液7.5 mL。
【步骤】在100 mL三颈烧瓶中分别安装搅拌器、温度计和冷凝管,依次加入1.3 g 氢氧化钠、12 mL水和7.5 mL。
95%乙醇。
搅拌混合物,使氢氧化钠溶解,待稍冷后加入3.4 mL对甲苯乙酮。
继续搅拌,从冷凝管上口缓慢滴加2.6 mL 苯甲醛到三颈烧瓶中,控制反应温度25~30℃。
加完后,继续搅拌2.5~3 h,并维持上述温度不变。
在反应后期,瓶内可能会有固体析出。
反应结束后,用冰浴冷却三颈烧瓶,并继续搅拌至有大量固体析出。
抽滤,用水洗涤结晶至滤液呈中性,然后用少量冰水浴冷却过的95%乙醇进行洗涤。
抽紧,压干,得粗产物4.8 g。
若要得到更纯的产物,可用95%乙醇进行重结晶,干燥后可得产物约4.0 g,熔点72~73℃。
纯1一对甲苯基一3一苯基一2一丙烯一1一酮的熔点为75℃。
本实验约需6 h。
【注释】[1]对甲苯乙酮的合成见实验二。
[2]苯甲醛使用前要新蒸馏过。
[3]由于反应放热,必要时可用冷水浴冷却。
[4]用95%乙醇溶液洗涤是为了除去未完全反应的对甲苯乙酮和苯甲醛。
【思考题】1.本实验的Claisen—Schmidt缩合反应中,可能会发生哪些副反应?它们是如何被抑制的?2.说明该反应以E型产物为主的原因。
除熔点外,还有哪些方法可以区别这两种顺反异构体?。

一、研究有机化合物的基本步骤常用的分离、提纯方法包括蒸馏、萃取、重结晶。
二、蒸馏1.蒸馏原理:利用有机物与杂质的沸点差异,将有机化合物以蒸汽的形式蒸出,然后冷凝得到产品。
2.适用对象:互相溶解、沸点不同的液态有机混合物3.适用条件:①有机物的热稳定性较强;②有机物与杂质的沸点相差较大(一般约大于30 ℃)4.实验仪器:铁架台、酒精灯、石棉网、蒸馏烧瓶、温度计、直形冷凝管、牛角管(尾接管)、锥形瓶。
5.实验装置与注意事项①蒸馏烧瓶里盛液体的用量不超2/3,不少于1/3; ②加入沸石或碎瓷片,防止暴沸;③温度计水银球应与蒸馏烧瓶的支管口平齐; ④冷凝水应下口进入,上口流出;⑤实验开始时,先通冷凝水水,后加热;实验结束时,先停止加热,后停止通冷凝水;第03讲 有机物的分离、提纯知识导航知识精讲三、萃取1.原理:(1)液—液萃取:利用待分离组分在两种不互溶的溶剂中的溶解性不同,使待分离组分从溶解度较小的溶剂中转移到溶解度较大的溶剂中。
(2)固—液萃取:用溶剂从固体物质中溶解出待分离组分。
2.萃取剂(1)选择原则①与原溶剂互不相溶;②与溶质、原溶剂均不反应;③溶质在萃取剂中的溶解度远大于原溶剂。
(2)常用萃取剂乙醚(C2H5OC2H5)、乙酸乙酯、二氯甲烷等3.分液:将萃取后的两层液体(互不相溶、密度也不同的两种液体)分离开的操作方法。
4.主要仪器:分液漏斗5.实验装置与注意事项①分液漏斗使用之前必须检漏(在分液漏斗中注入少量的水,塞上玻璃塞,倒置,看是否漏水,若不漏水,正立分液漏斗后将玻璃塞旋转180°,再倒置看是否漏水)。
②使用时需将漏斗上口的玻璃塞打开,或使玻璃塞上的凹槽对准分液漏斗上的小孔。
③漏斗下端管口紧靠烧怀内壁,分液时下层液体从下口流出,上层液体从上口倒出。
四、重结晶1.重结晶原理:利用被提纯物质与杂质在同一溶剂中的溶解度不同而将杂质除去。
2.适用对象:固体有机化合物3.溶剂选择:要求杂质在此溶剂中溶解度很小或溶解度很大,易于除去;被提纯的有机化合物在此溶剂中的溶解度受温度的影响较大,能够进行冷却结晶。


利用傅-克酰基化反应制备芳酮实验的改进秦丙昌;付小普;樊新衡【摘要】对傅-克酰基化反应的后处理步骤做了重要改进:分解芳酮-三氯化铝络合物时,不加盐酸,仅用水;粗产物在蒸馏前不再加固体干燥剂进行干燥,利用苯或甲苯能与水形成共沸物的性质将粗产物中的水带出.改进后的方法能节约试剂,节省时间,减少污染,提高产率.【期刊名称】《大学化学》【年(卷),期】2006(021)001【总页数】4页(P44-46,48)【关键词】傅-克酰基化反应;芳酮;实验;制备;三氯化铝;减少污染;干燥剂;络合物;后处理;水带【作者】秦丙昌;付小普;樊新衡【作者单位】安阳师范学院化学系,安阳,455002;安阳师范学院化学系,安阳,455002;安阳师范学院化学系,安阳,455002【正文语种】中文【中图分类】O61 问题的提出Friedel-Craftz酰基化反应,简称傅-克酰基化反应,是制备芳酮最重要和最常用的方法,在化工生产上被广泛应用,也是大学有机化学实验的重要内容。
国内出版的有机化学实验教材中,通常安排了利用傅-克酰基化反应制备苯乙酮[1,2,3]、对甲苯乙酮[4]或二苯酮[1,4]等实验内容,操作步骤基本相同:在无水条件下,用无水三氯化铝做催化剂,使酰基化试剂(乙酐或苯甲酰氯)与过量芳烃(苯或甲苯)在一定温度或回流状态下反应至无氯化氢气体逸出为止。
然后将反应混合物倒入冰水和浓盐酸的混合物中,或者在冷却和搅拌下将冰水和浓盐酸的混合物滴入反应瓶中,分解芳酮-三氯化铝络合物,使酮游离出来。
沉淀溶解后,依次进行萃取(苯乙酮的制备)、洗涤、干燥、蒸馏,得到产物。
在上述实验过程中,反应完成后进行后处理的一个关键步骤是用冰水和浓盐酸的混合物分解芳酮-三氯化铝络合物。
那么,在此过程中为什么要加盐酸呢?一般认为,三氯化铝与水反应会生成氢氧化铝沉淀[2,5]:AlCl3+3H2OAl(OH)3↓+3HCl需要加入盐酸使氢氧化铝沉淀溶解。
〔19〕中华人民共和国专利局〔12〕发明专利申请公开说明书[11]公开号CN 1072925A〔43〕公开日1993年6月9日[21]申请号91111452.1[22]申请日91.12.3[71]申请人陈敏哲地址132000吉林省吉林市吉林造纸厂宅24号楼96号[72]发明人陈敏哲 [51]Int.CI 5C07C 63/30C07C 49/213C07C 51/58权利要求书 1 页 说明书 3 页 附图 1 页[54]发明名称联体法生产对苯二甲酰氯、间苯二甲酰氯和苯乙酮[57]摘要本发明公布了一种用二甲苯联体生产对苯二甲酰氯、间苯二甲酰氯、苯乙酮的新方法。
其特点在于该方法通过二甲苯氯化,水解后而得对苯二甲酰氯、间苯二甲酰氯、苯乙酮及副产物邻羧基苯甲醛的混合物,然后使其分离而得目的产物对苯二甲酰氯、间苯二甲酰氯、苯乙酮。
该方法克服以往诸路线工序长、成本高、原料昂贵的弊病,以技术新、投资少、效益高等特点开辟了一条联体生产的新路线。
91111452.1权 利 要 求 书第1/1页1、用二甲苯联体生产对苯二甲酰氯,间苯二甲酰氯、苯乙酮的新路线。
其特征是二甲苯不经分离即进行混合侧链氯化。
然后采用精馏方法进行产品分离。
2、根据权利要求1所规定的路线,进行混合氯化所用的二甲苯中对二甲苯、间二甲苯、乙苯和邻二甲苯之比例可以是任意的,包括使用单一的纯组分和使用缺一至二种组分的混合物。
3、根据按权利要求1所规定的路线其特征是用水解方法使氯化混合物生成对苯二甲酰氯,间苯二甲酰氯、苯乙酮及邻羧基苯甲醛,其被水解的氯化混合物比例是任意的。
4、按权利要求1所规定的路线,采用减压精馏方式,将混合水解产物分离成对苯二甲酰氯、间苯二甲酰氯、苯乙酮及副产物邻羧基苯甲醛。
91111452.1说 明 书第1/3页联体法生产对苯二甲酰氯、间苯二甲酰氯和苯乙酮本发明涉及一种用二甲苯联体生产对苯二甲酰氯、间苯二甲酰氯和苯乙酮的新路线。
原有生产苯乙酮的技术是:苯和乙酰氯在三氯化铝存在下经缩合反应而制得,或苯与醋酐经缩合而制得,原有生产对苯二甲酰氯和间苯二甲酰氯技术均未见报导。
(19)中华人民共和国国家知识产权局(12)发明专利申请(10)申请公布号 (43)申请公布日 (21)申请号 201811090933.9(22)申请日 2018.09.19(71)申请人 江苏扬农化工集团有限公司地址 225009 江苏省扬州市文峰路39号申请人 江苏瑞祥化工有限公司 宁夏瑞泰科技股份有限公司(72)发明人 朱叶峰 丁克鸿 徐林 王怡明 孙伟 顾峰 刘补娥 (74)专利代理机构 北京恒和顿知识产权代理有限公司 11014代理人 揭玉斌(51)Int.Cl.C07D 233/90(2006.01)(54)发明名称一种氰霜唑的制备方法(57)摘要本发明公开了一种氰霜唑的制备方法,包括:(1)5-(对甲苯基)-1H -咪唑、无水碳酸钾、乙酸乙酯和N,N -二甲胺基磺酰氯,50℃-77℃反应6h -12h,之后降至室温,水洗,萃取,干燥,蒸除乙酸乙酯得中间体A粗品,再重结晶得中间体A;(2)中间体A、铜盐、氰基化试剂、氧化剂以及N ,N –二甲基甲酰胺,110℃-130℃反应6h -12h,之后降至室温,回收N ,N –二甲基甲酰胺,水洗,萃取,干燥,蒸除乙酸乙酯得中间体B粗品,再重结晶得中间体B;(3)中间体B、N -氯代丁二酰亚胺以及乙腈,室温下搅拌反应2h -6h,反应结束回收溶剂乙腈,加入碳酸氢钠溶液中和至中性,萃取,干燥,蒸除乙酸乙酯得氰霜唑粗品,再重结晶得氰霜唑。
本发明反应收率高,选择性好。
权利要求书1页 说明书4页CN 108912052 A 2018.11.30C N 108912052A1.一种氰霜唑的制备方法,所述方法的反应式如下:所述制备方法包括:(1)向反应瓶中加入5-(对甲苯基)-1H -咪唑、无水碳酸钾、乙酸乙酯和N,N -二甲胺基磺酰氯,升温至50℃-77℃反应6h -12h,反应结束后降温至室温,加入水洗涤后,再用乙酸乙酯萃取,无水硫酸钠干燥,蒸除乙酸乙酯得到中间体A粗品,所述中间体A粗品用乙酸乙酯重结晶后得到中间体A;(2)向反应瓶中加入所述中间体A、铜盐、氰基化试剂、氧化剂以及N ,N –二甲基甲酰胺,升温至110℃-130℃反应6h -12h,反应结束后降温至室温,回收溶剂N ,N –二甲基甲酰胺,加入水洗,乙酸乙酯萃取,无水硫酸钠干燥,蒸除乙酸乙酯得到中间体B粗品,所述中间体B粗品用乙酸乙酯重结晶得到中间体B;所述铜盐选自碘化亚铜、氯化亚铜、硫酸铜、乙酸铜、三氟乙酸铜以及氯化铜中一种或多种;所述氰基化试剂为氰化钾、氰化钠或者氰基三甲基硅烷;所述氧化剂为过硫酸钾、N -氟代双苯磺酰胺或者苯醌;(3)向反应瓶中加入所述中间体B、N -氯代丁二酰亚胺以及乙腈,室温下搅拌反应2h -6h,反应结束回收溶剂乙腈,加入浓度为20%的碳酸氢钠溶液中和至中性,乙酸乙酯萃取,无水硫酸钠干燥,蒸除乙酸乙酯得到氰霜唑粗品,所述氰霜唑粗品用乙酸乙酯重结晶得到白色固体氰霜唑。