国家食品药品监督管理总局通告2014年第8号 关于发布第一类医疗器械产品目录的通告
- 格式:pdf
- 大小:435.32 KB
- 文档页数:61

医疗器械:医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。
医疗机械分类目录:一、新《分类目录》的总体说明:(一)新《分类目录》按技术专业和临床使用特点分为22个子目录,子目录由一级产品类别、二级产品类别、产品描述、预期用途、品名举例和管理类别组成。
判定产品的管理类别时,应当根据产品的实际情况,结合新《分类目录》中产品描述、预期用途和品名举例进行综合判定,产品描述和预期用途是用于判定产品的管理类别,不代表相关产品注册内容的完整表述。
(二)新《分类目录》不包括体外诊断试剂,体外诊断试剂产品类别应当按照《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号,以下简称5号令)、《体外诊断试剂注册管理办法修正案》(总局令第30号,以下简称30号令)、《6840体外诊断试剂分类子目录(2013版)》及后续发布的分类界定文件中有关体外诊断试剂的分类界定意见进行判定,分类编码继续延用6840。
(三)新《分类目录》不包括组合包类产品,组合包类产品的类别应当依据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、5号令、30号令等相关规定进行判定。
(四)《关于发布第一类医疗器械产品目录的通告》(国家食品药品监督管理总局通告2014年第8号)、《食品药品监管总局办公厅关于实施第一类医疗器械备案有关事项的通知》(食药监办械管〔2014〕174号)和2014年5月30日以后发布的医疗器械分类界定文件中有关第一类医疗器械产品的分类界定意见继续有效。
自2018年8月1日起,上述文件规定的产品管理类别与新《分类目录》不一致的,以新《分类目录》的产品管理类别为准。
(五)自2018年8月1日起,除第(二)项和第(四)项以及既往发布的分类界定文件中不作为医疗器械管理的产品分类界定意见外,原《医疗器械分类目录》(国药监械〔2002〕302号,以下简称原《分类目录》)及既往发布的医疗器械分类界定文件内容及目录废止。

《医疗器械监督管理条例》于2014年6月1日起正式实施,并于2017年5月4日修订,各类配套规章及规范性文件亦于近年来陆续制修订并发布。
各医疗器械企业作为受法规变化影响最大的相关方,应积极应对新法规政策下的挑战,密切关注最新法规动态,提前做好攻略规划,确保产品尽快获批上市!完善自身法规体系,确保企业合法合规生产经营!
为方便各企业检索最新法规,本协会特收集、整理、编制国家药品监督管理局(NMPA)已发布的医疗器械法规文件(包括但不限于)清单,供大家参考。
各企业应根据自身实际情况及产品领域,对各类法规进行识别、收集、评估、导入、宣贯培训。
注:未经许可,严禁转载!
若需转载,请注明出处:来源于深圳市医疗器械行业协会!
一、行政法规
二、部门规章
三、通告
四、公告
五、通知性文件
六、政策解读
七、相关法律法规
深圳市医疗器械行业协会
2020年1月。

国家食品药品监督管理总局公告2014年第8号――药
品GMP认证公告(第18号)
文章属性
•【制定机关】国家食品药品监督管理总局(已撤销)
•【公布日期】2014.03.06
•【文号】国家食品药品监督管理总局公告2014年第8号
•【施行日期】2014.03.06
•【效力等级】部门规范性文件
•【时效性】现行有效
•【主题分类】药政管理
正文
国家食品药品监督管理总局公告
(2014年第8号)
药品GMP认证公告(第18号)
按照《药品生产质量管理规范认证管理办法》的规定,经现场检查和审核批准,苏州中化药品工业有限公司等43家药品生产企业符合《药品生产质量管理规范(2010年修订)》要求,发给《药品GMP证书》。
特此公告。
附件:药品GMP认证目录(第18号)
国家食品药品监督管理总局
2014年3月6日附件:
药品GMP认证目录(第18号)。

国家食品药品监督管理总局令第 8 号《医疗器械经营监督管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。
局长张勇2014年7月30日医疗器械经营监督管理办法第一章总则第一条为加强医疗器械经营监督管理,规范医疗器械经营行为,保证医疗器械安全、有效,根据《医疗器械监督管理条例》,制定本办法。
第二条在中华人民共和国境内从事医疗器械经营活动及其监督管理,应当遵守本办法。
第三条国家食品药品监督管理总局负责全国医疗器械经营监督管理工作。
县级以上食品药品监督管理部门负责本行政区域的医疗器械经营监督管理工作。
上级食品药品监督管理部门负责指导和监督下级食品药品监督管理部门开展医疗器械经营监督管理工作。
第四条按照医疗器械风险程度,医疗器械经营实施分类管理。
经营第一类医疗器械不需许可和备案,经营第二类医疗器械实行备案管理,经营第三类医疗器械实行许可管理。
第五条国家食品药品监督管理总局制定医疗器械经营质量管理规范并监督实施。
第六条食品药品监督管理部门依法及时公布医疗器械经营许可和备案信息。
申请人可以查询审批进度和审批结果,公众可以查阅审批结果。
第二章经营许可与备案管理第七条从事医疗器械经营,应当具备以下条件:(一)具有与经营范围和经营规模相适应的质量管理机构或者质量管理人员,质量管理人员应当具有国家认可的相关专业学历或者职称;(二)具有与经营范围和经营规模相适应的经营、贮存场所;(三)具有与经营范围和经营规模相适应的贮存条件,全部委托其他医疗器械经营企业贮存的可以不设立库房;(四)具有与经营的医疗器械相适应的质量管理制度;(五)具备与经营的医疗器械相适应的专业指导、技术培训和售后服务的能力,或者约定由相关机构提供技术支持。
从事第三类医疗器械经营的企业还应当具有符合医疗器械经营质量管理要求的计算机信息管理系统,保证经营的产品可追溯。
鼓励从事第一类、第二类医疗器械经营的企业建立符合医疗器械经营质量管理要求的计算机信息管理系统。


第一类医疗器械(含第一类体外诊断试剂)备案办理须知办事依据:《医疗器械监督管理条例》(国务院令第650号)、《关于第一类医疗器械备案有关事项的公告》(国家食品药品监督管理总局2014年第26号公告)、《国家食品药品监督管理总局关于发布第一类医疗器械产品目录的通告》(第8号)、《国家食品药品监督管理总局关于发布医疗器械产品技术要求编写指导原则的通告》(第9号)、《关于医疗器械生产经营备案有关事项的通知》(烟食药监械〔2014〕71号)瞩怂润厉钐瘗睐枥庑赖赁轫胧。
资料要求及说明备案资料第一类医疗器械备案表4.5.(1,目(2(3(4(5(6)同类产品不良事件情况说明。
6.产品说明书及最小销售单元标签设计样稿医疗器械应符合相应法规规定。
体外诊断试剂产品应按照《体外诊断试剂说明书编写指导原则》的有关要求,并参考有关技术指导原则编写产品说明书。
茕桢广鳓鲱选块网羁泪镀齐钧。
7.生产制造信息对生产过程相关情况的概述。
无源医疗器械应明确产品生产加工工艺,注明关键工艺和特殊工艺。
有源医疗器械应提供产品生产工艺过程的描述性资料,可采用流程图的形式,是生产过程的概述。
体外诊断试剂应概述主要生产工艺,包括:固相载体、显色系统等的描述及确定依据,反应体系包括样本采集及处理、样本要求、样本用量、试剂用量、反应条件、校准方法(如果需要)、质控方法等。
鹅娅尽损鹌惨历茏鸳赖萦诘聋。
应概述研制、生产场地的实际情况。
8.证明性文件企业营业执照复印件、组织机构代码证复印件。
9.符合性声明(1)声明符合医疗器械备案相关要求;(2)声明本产品符合第一类医疗器械产品目录或相应体外诊断试剂分类子目录的有关内容;(3)声明本产品符合现行国家标准、行业标准并提供符合标准的清单;(4)声明所提交备案资料的真实性。
(二)变更备案资料1.变化情况说明及相关证明文件第8号)2.3.(1(2(3(4(二)五、办理机构烟台市政务服务中心地址:烟台市芝罘区菜市街46号六、收费标准本项目不收费七、下载表格1.第一类医疗器械备案表2.备案信息表变化内容比对列表备案号:第一类医疗器械备案表产品名称(产品分类名称):备案人:烟台市食品药品监督管理局制填表说明本表用于境内第一类医疗器械、体外诊断试剂备案。

中国医疗器械法规清单(更新至2020/01)《医疗器械监督管理条例》于2014年6月1日起正式实施,并于2017年5月4日修订,各类配套规章及规范性文件亦于近年来陆续制修订并发布。
各医疗器械企业作为受法规变化影响最大的相关方,应积极应对新法规政策下的挑战,密切关注最新法规动态,提前做好攻略规划,确保产品尽快获批上市!完善自身法规体系,确保企业合法合规生产经营!为方便各企业检索最新法规,本协会特收集、整理、编制国家药品监督管理局(NMPA)已发布的医疗器械法规文件(包括但不限于)清单,供大家参考。
各企业应根据自身实际情况及产品领域,对各类法规进行识别、收集、评估、导入、宣贯培训。
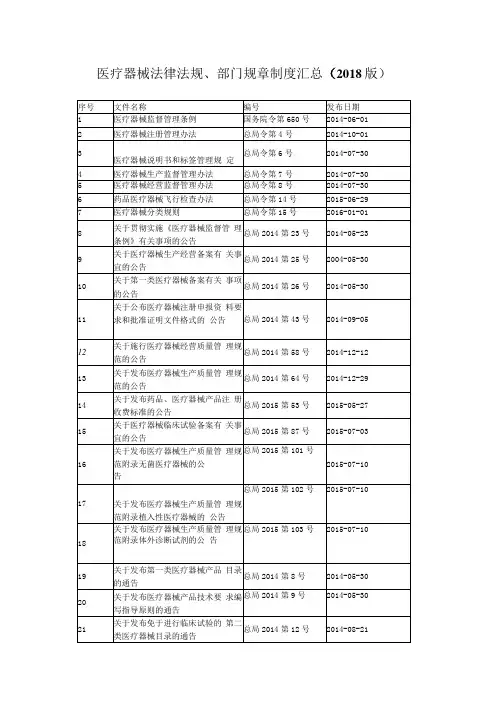
一、行政法规1. 《医疗器械监督管理条例》(国务院令第680号)二、部门规章1. 医疗器械注册管理办法(CFDA局令第4号)2. 体外诊断试剂注册管理办法(CFDA局令第5号)3. 医疗器械说明书和标签管理规定(CFDA局令第6号)4. 医疗器械生产监督管理办法(CFDA局令第7号)5. 医疗器械经营监督管理办法(CFDA局令第8号)6. 药品医疗器械飞行检查办法(CFDA局令第14号)7. 医疗器械分类规则(CFDA局令第15号)8. 医疗器械使用质量监督管理办法(CFDA局令第18号)9. 医疗器械通用名称命名规则(CFDA局令第19号)10. 医疗器械临床试验质量管理规范(CFDA国家卫计委令第25号)11. 医疗器械召回管理办法(CFDA局令第29号)12. 体外诊断试剂注册管理办法修正案(CFDA局令第30号)13. 关于调整部分医疗器械行政审批事项审批程序的决定(CFDA局令第32号)14. 医疗器械标准管理办法(CFDA局令第33号)15. 医疗器械网络销售监督管理办法(CFDA局令第38号)16. 医疗器械不良事件监测和再评价管理办法(国家市场监督管理总局令第1号)三、通告1. 关于发布第一类医疗器械产品目录的通告(CFDA通告2014年第8号)2. 关于发布医疗器械产品技术要求编写指导原则的通告(CFDA通告2014年第9号)3. 关于发布需进行临床试验审批的第三类医疗器械目录的通告(CFDA通告2014年第14号)4. 关于发布体外诊断试剂临床试验技术指导原则的通告(CFDA通告2014年第16号)5. 关于发布体外诊断试剂说明书编写指导原则的通告(CFDA通告2014年第17号)6. 关于发布禁止委托生产医疗器械目录的通告(CFDA 通告2014年第18号)7. 关于发布医疗器械生产企业供应商审核指南的通告(CFDA通告2015年第1号)8. 关于发布医疗器械临床评价技术指导原则的通告(CFDA通告2015年第14号)9. 关于发布医疗器械产品出口销售证明管理规定的通告(CFDA通告2015年第18号)10. 关于贯彻落实小微企业行政事业性收费优惠政策的通告(CFDA通告2015年第31号)11. 关于生产一次性使用无菌注、输器具产品有关事项的通告(CFDA通告2015年第71号)12. 关于发布医疗器械注册证补办程序等5个相关工作程序的通告(CFDA通告2015年第91号)13. 关于发布医疗器械注册指定检验工作管理规定的通告(CFDA通告2015年第94号)14. 关于发布医疗器械工艺用水质量管理指南的通告(CFDA通告2016年第14号)15. 关于发布《医疗器械临床试验伦理审查申请与审批表范本》等六个文件的通告(CFDA通告2016年第58号)16. 关于发布医疗器械生产企业质量管理体系年度自查报告编写指南的通告(CFDA通告2016年第76号)17. 关于发布医疗器械生产企业质量控制与成品放行指南的通告(CFDA通告2016年第173号)18. 关于发布医疗器械网络安全注册技术审查指导原则的通告(CFDA通告2017年第13号)19. 关于发布医疗器械审评沟通交流管理办法(试行)的通告(CFDA通告2017年第19号)20. 关于发布医疗器械优先审批申报资料编写指南(试行)的通告(CFDA通告2017年第28号)21. 关于实施《医疗器械分类目录》有关事项的通告(CFDA通告2017年第143号)22. 关于发布免于进行临床试验的体外诊断试剂临床评价资料基本要求(试行)的通告(CFDA通告2017年第179号)23. 关于需审批的医疗器械临床试验申请沟通交流有关事项的通告(CFDA通告2017年第184号)24. 关于发布医疗器械注册单元划分指导原则的通告(CFDA通告2017年第187号)25. 关于发布移动医疗器械注册技术审查指导原则的通告(CFDA通告2017年第222号)26. 关于过敏原类、流式细胞仪配套用、免疫组化和原位杂交类体外诊断试剂产品属性及类别调整的通告(CFDA通告2017年第226号)27. 关于发布接受医疗器械境外临床试验数据技术指导原则的通告(CFDA通告2018年第13号)28. 关于公布新修订免于进行临床试验医疗器械目录的通告(NMPA通告2018年第94号)29. 关于发布医疗器械生产企业管理者代表管理指南的通告(NMPA通告2018年第96号)30. 关于医疗器械经营企业跨行政区域设置库房办理事项的通告(NMPA通告2018年第108号)31. 关于发布创新医疗器械特别审查申报资料编写指南的通告(NMPA通告2018年第127号)32. 关于调整药械组合产品属性界定有关事项的通告(NMPA通告2019年第28号)33. 关于发布医疗器械注册申请电子提交技术指南的通告(NMPA通告2019年第29号)34. 关于医疗器械电子申报有关资料要求的通告(NMPA 通告2019年第41号)35. 关于发布《医疗器械产品注册项目立卷审查要求(试行)》等文件的通告(NMPA通告2019年第42号)36. 关于发布医疗器械生产质量管理规范附录独立软件的通告(NMPA通告2019年第43号)37. 关于做好第一批实施医疗器械唯一标识工作有关事项的通告(NMPA通告2019年第72号)38. 关于公布新增和修订的免于进行临床试验医疗器械目录的通告(NMPA通告2019年第91号)39. 关于发布医疗器械附条件批准上市指导原则的通告(NMPA通告2019年第93号)40. 关于发布医疗器械通用名称命名指导原则的通告(NMPA通告2019年第99号)四、公告1. 关于医疗器械生产经营备案有关事宜的公告(CFDA 公告2014年第25号)2. 关于第一类医疗器械备案有关事项的公告(CFDA公告2014年第26号)3. 关于公布医疗器械注册申报资料要求和批准证明文件格式的公告(CFDA公告2014年第43号)4. 关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告(CFDA公告2014年第44号)5. 关于施行医疗器械经营质量管理规范的公告(CFDA 公告2014年第58号)6. 关于发布医疗器械生产质量管理规范的公告(CFDA 公告2014年第64号)7. 关于发布药品、医疗器械产品注册收费标准的公告(CFDA公告2015年第53号)8. 关于医疗器械临床试验备案有关事宜的公告(CFDA 公告2015年第87号)9. 关于发布医疗器械生产质量管理规范附录无菌医疗器械的公告(CFDA公告2015年第101号)10. 关于发布医疗器械生产质量管理规范附录植入性医疗器械的公告(CFDA公告2015年第102号)11. 关于发布医疗器械生产质量管理规范附录体外诊断试剂的公告(CFDA公告2015年第103号)12. 关于境内医疗器械生产企业跨省新开办企业时办理产品注册及生产许可有关事宜的公告(CFDA公告2015年第203号)13. 关于规范含银盐医疗器械注册管理有关事宜的公告(CFDA公告2015年第225号)14. 关于发布医疗器械冷链(运输、贮存)管理指南的公告(CFDA公告2016年第154号)15. 关于发布医疗器械优先审批程序的公告(CFDA公告2016年第168号)16. 关于发布医疗器械生产质量管理规范附录定制式义齿的公告(CFDA公告2016年第195号)17. 关于发布医疗器械技术审评专家咨询委员会管理办法的公告(CFDA公告2017年第36号)18. 关于第二批规范性文件清理结果的公告(CFDA公告2017年第88号)19. 关于发布医疗器械分类目录的公告(CFDA公告2017年第104号)20. 关于医疗器械经营备案有关事宜的公告(CFDA公告2017年第129号)21. 关于进口医疗器械注册申请人和备案人名称使用中文的公告(CFDA公告2017年第131号)22. 关于发布医疗器械临床试验机构条件和备案管理办法的公告(CFDA公告2017年第145号)23. 关于发布医疗器械标准制修订工作管理规范的公告(CFDA公告2017年第156号)24. 关于修改医疗器械延续注册等部分申报资料要求的公告(NMPA公告2018年第53号)25. 关于医疗器械规范性文件(1998—2013年)清理结果的公告(NMPA公告2018年第37号)26. 关于发布创新医疗器械特别审查程序的公告(NMPA 公告2018年第83号)27. 关于发布药品医疗器械境外检查管理规定的公告(NMPA公告2018年第101号)28. 关于调整医疗器械临床试验审批程序的公告(NMPA 公告2019年第26号)29. 关于实施医疗器械注册电子申报的公告(NMPA公告2019年第46号)30. 关于发布定制式医疗器械监督管理规定(试行)的公告(NMPA公告2019年第53号)31. 关于发布医疗器械唯一标识系统规则的公告(NMPA 公告2019年第66号)32. 关于修改一次性使用无菌导尿管(包)说明书等有关内容的公告(NMPA公告2019年第94号)五、通知性文件1. 关于印发医疗器械质量监督抽查检验管理规定的通知(食药监械监〔2013〕212号)2. 关于印发医疗器械生产日常监督现场检查工作指南的通知(食药监办械监〔2014〕7号)3. 关于实施《医疗器械生产监督管理办法》和《医疗器械经营监督管理办法》有关事项的通知(食药监械监〔2014〕143号)4. 关于实施《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》有关事项的通知(食药监械管〔2014〕144号)5. 关于实施第一类医疗器械备案有关事项的通知(食药监办械管〔2014〕174号)6. 关于印发医疗器械检验机构开展医疗器械产品技术要求预评价工作规定的通知(食药监械管〔2014〕192号)7. 关于印发境内第三类和进口医疗器械注册审批操作规范的通知(食药监械管〔2014〕208号)8. 关于印发境内第二类医疗器械注册审批操作规范的通知(食药监械管〔2014〕209号)9. 关于印发医疗器械生产企业分类分级监督管理规定的通知(食药监械监〔2014〕234号)10. 关于印发国家重点监管医疗器械目录的通知(食药监械监〔2014〕235号)11. 关于启用医疗器械生产经营许可备案信息系统的通知(食药监办械监函〔2014〕476号)12. 关于印发境内第三类医疗器械注册质量管理体系核查工作程序(暂行)的通知(食药监械管〔2015〕63号)13. 关于印发医疗器械经营企业分类分级监督管理规定的通知(食药监械监〔2015〕158号)14. 关于印发医疗器械经营环节重点监管目录及现场检查重点内容的通知(食药监械监〔2015〕159号)15. 关于印发医疗器械生产质量管理规范现场检查指导原则等4个指导原则的通知(食药监械监〔2015〕218号)16. 关于印发医疗器械经营质量管理规范现场检查指导原则的通知(食药监械监〔2015〕239号)17. 关于执行医疗器械和体外诊断试剂注册管理办法有关问题的通知(食药监械管〔2015〕247号)18. 关于印发医疗器械检验机构资质认定条件的通知(食药监科〔2015〕249号)19. 关于成立医疗器械分类技术委员会的通知(食药监械管〔2015〕259号)20. 关于启用医疗器械注册管理信息系统备案子系统的通知(食药监办械管函〔2015〕534号)21. 关于启用医疗器械注册管理信息系统受理和制证、技术审评、行政审批子系统的通知(食药监办械管函〔2015〕804号)22. 关于医疗器械产品技术要求有关问题的通知(食药监办械管〔2016〕22号)23. 关于实施《医疗器械通用名称命名规则》有关事项的通知(食药监械管〔2016〕35号)24. 关于印发一次性使用无菌注射器等25种医疗器械生产环节风险清单和检查要点的通知(食药监械监〔2016〕37号)25. 关于及时公开第二类医疗器械注册信息和第一类医疗器械产品备案信息的通知(食药监办械管〔2016〕65号)26. 关于体外诊断试剂说明书文字性变更有关问题的通知(食药监办械管〔2016〕117号)27. 关于印发医疗器械生产质量管理规范定制式义齿现场检查指导原则的通知(食药监械监〔2016〕165号)28. 关于印发一次性使用塑料血袋等21种医疗器械生产环节风险清单和检查要点的通知(食药监械监〔2017〕14号)29. 关于第一类、第二类医疗器械生产企业实施医疗器械生产质量管理规范有关工作的通知(食药监办械监〔2017〕120号)30. 关于规范医疗器械产品分类有关工作的通知(食药监办械管〔2017〕127号)31. 关于做好医疗器械临床试验机构备案工作的通知(食药监办械管〔2017〕161号)32. 关于做好医疗器械检验有关工作的通知(食药监办械管〔2017〕187号)33. 关于实施《医疗器械网络销售监督管理办法》有关事项的通知(食药监办械监〔2018〕31号)34. 关于印发医疗器械注册技术审查指导原则制修订工作管理规范的通知(药监办〔2018〕13号)35. 关于加强医疗器械生产经营许可(备案)信息管理有关工作的通知(2018年08月02日发布)36. 关于贯彻落实国务院“证照分离”改革要求做好医疗器械上市后监管审批相关工作的通知(药监综械管〔2018〕39号)37. 关于贯彻落实“证照分离”改革措施进一步推进医疗器械审评审批制度改革的通知(药监综械注〔2018〕43号)38. 关于印发医疗器械临床试验检查要点及判定原则的通知(药监综械注〔2018〕45号)39. 关于扩大医疗器械注册人制度试点工作的通知(国药监械注〔2019〕33号)40. 关于印发医疗器械检验工作规范的通知(国药监科外〔2019〕41号)41. 关于印发医疗器械唯一标识系统试点工作方案的通知(药监综械注〔2019〕56号)42. 2018年医疗器械产品分类界定结果汇总(2019年02月18日发布)43. 2019年第一批医疗器械产品分类界定结果汇总(2019年07月18日发布)44. 2019年第二批医疗器械产品分类界定结果汇总(2019年11月25日发布)六、政策解读1. 《医疗器械经营监督管理办法》部分(一)(2015年01月22日发布)2. 《医疗器械生产监督管理办法》部分(一)(2015年01月22日发布)3. 医疗器械注册管理法规解读之一(《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》部分)(2015年02月05日发布)4. 医疗器械注册管理法规解读之二(《医疗器械说明书和标签管理规定》部分)(2015年02月05日发布)5. 关于《药品医疗器械飞行检查办法》的说明(2015年07月08日发布)6. 医疗器械注册管理法规解读之三(关于《医疗器械分类规则》的修订说明)(2015年07月16日发布)7. 医疗器械注册管理法规解读之四(2015年11月02日发布)8. 医疗器械注册管理法规解读之五(2015年11月19日发布)9. 关于《医疗器械使用质量监督管理办法》的说明(2015年11月30日发布)10. 医疗器械注册管理法规解读之六(2016年01月07日发布)11. 关于《医疗器械通用名称命名规则》的说明(2016年01月27日发布)12. 《医疗器械临床试验质量管理规范》解读(2016年03月23日发布)13. 关于《医疗器械优先审批程序》的说明(2016年10月26日发布)14. YY/T0287-2017idt ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准解读(一)(2017年02月04日发布)15. YY/T0287-2017idt ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准解读(二)(2017年02月04日发布)16. 《医疗器械召回管理办法》解读(2017年02月08日发布)17. 《体外诊断试剂注册管理办法修正案》解读(2017年02月08日发布)18. 《医疗器械网络安全注册技术审查指导原则》解读(2017年03月02日发布)19. 《医疗器械标准管理办法》解读(2017年04月26日发布)20. 《医疗器械召回管理办法》解读之二(2017年05月24日发布)21. 图解政策:《医疗器械召回管理办法》解读(2017年06月22日发布)22. 图解政策:医疗器械不良事件那些事(2017年06月24日发布)23. 图解政策:体外诊断试剂注册管理办法修正案(2017年07月28日发布)24. 创新医疗器械特别审批程序相关问题解读(2017年07月31日发布)25. 医疗器械临床试验质量管理相关问题解读(2017年07月31日发布)26. 《医疗器械标准管理办法》解读之一(2017年08月09日发布)27. 《医疗器械标准管理办法》解读之二(2017年08月09日发布)28. 《医疗器械网络安全注册技术审查指导原则》解读(2017年08月09日发布)29. 图解政策:医疗器械临床试验质量管理相关问题解读(2017年09月04日发布)30. 图解政策:创新医疗器械特别审批程序相关问题解读(2017年09月04日发布)31. 图解政策:ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准解读(一)(2017年09月12日发布)32. 图解政策:ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准解读(二)(2017年09月12日发布)33. 《免于进行临床试验的体外诊断试剂临床评价资料基本要求(试行)》解读(2017年11月08日发布)34. 《医疗器械临床试验机构条件和备案管理办法》解读(2017年11月24日发布)35. 《医疗器械网络销售监督管理办法》解读(2017年12月22日发布)36. 《移动医疗器械注册技术审查指导原则》解读(2017年12月29日发布)37. 图解政策:医疗器械网络销售监督管理办法解读一(2018年01月12日发布)38. 图解政策:医疗器械网络销售监督管理办法解读二(2018年01月15日发布)39. 医疗器械临床试验质量管理相关问题解读之二(2018年04月18日发布)40. 《医疗器械分类目录》实施有关问题解读(2018年08月01日发布)41. 图解政策:国家药品监督管理局办公室关于强脉冲光脱毛类产品分类界定的通知(2018年09月26日发布)42. 图解政策:医疗器械注册技术审查指导原则制修订工作管理规范(2018年09月28日发布)43. 图解政策:关于加强医疗器械生产经营许可(备案)信息管理有关工作的通知(2018年09月28日发布)44. 免于进行临床试验医疗器械目录解读(2018年09月30日发布)45. 图解政策:《医疗器械分类目录》实施有关问题解读(2018年10月09日发布)46. 图解政策:新修订的《免于进行临床试验医疗器械目录》解读(2018年10月19日发布)47. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之一(2018年10月24日发布)48. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之二(2018年10月25日发布)49. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之三(2018年10月26日发布)50. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之四(2018年10月29日发布)51. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之五(2018年10月30日发布)52. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之六(2018年10月31日发布)53. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之七(2018年11月01日发布)54. 图解政策:《医疗器械不良事件监测和再评价监督管理办法》之八(2018年11月02日发布)55. 《创新医疗器械特别审查程序》解读(2018年11月05日发布)56. 图解政策:医疗器械生产企业管理者代表管理指南之一(2018年11月06日发布)57. 图解政策:医疗器械生产企业管理者代表管理指南之二(2018年11月06日发布)58. 图解政策:医疗器械生产企业管理者代表管理指南之三(2018年11月07日发布)59. 图解政策:关于优化优先审评申请审核工作程序的通知(2018年11月14日发布)60. 图解政策:用于罕见病防治医疗器械注册审查指导原则(之一)(2018年11月29日发布)61. 图解政策:用于罕见病防治医疗器械注册审查指导原则(之二)(2018年11月30日发布)62. 图解政策:关于贯彻落实“证照分离”改革措施进一步推进医疗器械审评审批制度改革的通知(2018年12月03日发布)63. 图解政策:创新医疗器械特别审查程序之一(2018年12月14日发布)64. 图解政策:创新医疗器械特别审查程序之二(2018年12月19日发布)65. 图解政策:创新医疗器械特别审查程序之三(2018年12月19日发布)66. 图解政策:药品医疗器械境外检查管理规定(一)(2019年02月01日发布)67. 图解政策:药品医疗器械境外检查管理规定(二)(2019年02月12日发布)68. 《定制式医疗器械监督管理规定(试行)》解读(2019年07月04日发布)69. 《医疗器械唯一标识系统规则》解读(2019年08月27日发布)70. 免于进行临床试验医疗器械目录汇总(2019年12月23日发布)71. 图解政策:国家药监局关于发布医疗器械附条件批准上市指导原则的通告(2019年第93号)(一)(2020年01月02日发布)72. 图解政策:国家药监局关于发布医疗器械附条件批准上市指导原则的通告(2019年第93号)(二)(2020年01月02日发布)73. 图解政策:国家药监局关于发布医疗器械附条件批准上市指导原则的通告(2019年第93号)(三)(2020年01月02日发布)七、相关法律法规1. 中华人民共和国广告法(2018年11月05日发布)2. 药品、医疗器械、保健食品、特殊医学用途配方食品广告审查管理暂行办法(国家市场监督管理总局令第21号)。

医疗器械注册员必备法规《医疗器械监督管理条例》(国务院令第650号)自2014年6月1日正式实施,条例配套的规章及规范性文件陆续密集出台。
新法规的出台意味着医疗器械企业面临新的机遇和挑战,作为医疗器械注册人熟悉这些必备的法规,对企业正常运行、产品如期顺利上市起到至关重要的作用国家食品药品监督管理总局已经发布和正在征求意见的系列法规文件目录如下,具体内容参见国家食品药品监督管理总局网站。
行政法规1. 医疗器械监督管理条例(国务院令第650号)2. 国务院关于修改《医疗器械监督管理条例》的决定(国务院令第680号)部门规章1. 医疗器械注册管理办法(CFDA局令第4号)2. 体外诊断试剂注册管理办法(CFDA局令第5号)3. 医疗器械说明书和标签管理规定(CFDA局令第6号)4. 医疗器械生产监督管理办法(CFDA局令第7号)医疗器械生产监督管理办法(修正版)(2017年11月21日发布)5. 医疗器械经营监督管理办法(CFDA局令第8号)医疗器械经营监督管理办法(修正版)(2017年11月21日发布)6. 药品医疗器械飞行检查办法(CFDA局令第14号)7. 医疗器械分类规则(CFDA局令第15号)8. 医疗器械使用质量监督管理办法(CFDA局令第18号)9. 医疗器械通用名称命名规则(CFDA局令第19号)10. 医疗器械临床试验质量管理规范(CFDA 国家卫计委令第25号)11. 医疗器械召回管理办法(CFDA局令第29号)12. 体外诊断试剂注册管理办法修正案(CFDA局令第30号)13. 关于调整部分医疗器械行政审批事项审批程序的决定(CFDA局令第32号)14. 医疗器械标准管理办法(CFDA局令第33号)15. 医疗器械网络销售监督管理办法(CFDA局令第38号)产品分类界定1. 关于可降解泪道栓子等53个产品分类界定的通知(食药监办〔2013〕11号)2. 关于体外高频治疗机等47个产品分类界定的通知(食药监办械管〔2013〕31号)3. 关于血细胞分离机用耗材等11个产品分类界定的通知(食药监办械管〔2013〕68号)4. 关于自体富血小板凝胶制备用套装等23个产品分类界定的通知(食药监办械管〔2013〕69号)5. 关于重症及麻醉临床信息系统等9个产品分类界定的通知(食药监办械管〔2013〕109号)6. 关于基因分析仪等3个产品分类界定的通知(食药监办械管〔2014〕8号)7. 关于生物电导扫描仪等11个产品分类界定的通知(食药监办械管〔2014〕10号)8. 关于角膜治疗仪等12个产品分类界定的通知(食药监办械管〔2014〕103号)9. 关于电子宫腔观察镜等30个产品分类界定的通知(食药监办械管〔2014〕149号)10. 关于交联胺化聚乙烯醇泡沫封堵肺减容系统等34个产品分类界定的通知(食药监办械管〔2014〕177号)11. 关于腹腔镜手术用内窥镜自动调控定位装置等61个产品分类界定的通知(食药监办械管〔2014〕198号)12. 关于乳腺摄影立体定位装置等153个产品分类界定的通知(食药监办械管〔2015〕49号)13. 关于人工血管接环等172个产品分类界定的通知(食药监办械管〔2015〕69号)14. 关于恒温核酸扩增检测仪等22个产品分类界定的通知(食药监办械管〔2015〕75号)15. 关于多功能超声骨刀等127个产品分类界定的通知(食药监办械管〔2015〕104号)16. 关于阴茎增大增粗拉伸器具产品分类界定事项的复函(食药监办械管函〔2016〕480号)通告1. 关于发布第一类医疗器械产品目录的通告(CFDA通告2014年第8号)2. 关于发布医疗器械产品技术要求编写指导原则的通告(CFDA通告2014年第9号)3. 关于发布免于进行临床试验的第二类医疗器械目录的通告(CFDA通告2014年第12号)4. 关于发布免于进行临床试验的第三类医疗器械目录的通告(CFDA通告2014年第13号)5. 关于发布需进行临床试验审批的第三类医疗器械目录的通告(CFDA通告2014年第14号)6. 关于医疗器械生产质量管理规范执行有关事宜的通告(CFDA通告2014年第15号)7. 关于发布体外诊断试剂临床试验技术指导原则的通告(CFDA通告2014年第16号)8. 关于发布体外诊断试剂说明书编写指导原则的通告(CFDA通告2014年第17号)9. 关于发布禁止委托生产医疗器械目录的通告(CFDA通告2014年第18号)10. 关于发布医疗器械生产企业供应商审核指南的通告(CFDA通告2015年第1号)11. 关于发布医疗器械临床评价技术指导原则的通告(CFDA通告2015年第14号)12. 关于发布医疗器械产品出口销售证明管理规定的通告(CFDA通告2015年第18号)13. 关于贯彻落实小微企业行政事业性收费优惠政策的通告(CFDA通告2015年第31号)14. 关于生产一次性使用无菌注、输器具产品有关事项的通告(CFDA通告2015年第71号)15. 关于发布医疗器械注册证补办程序等5个相关工作程序的通告(CFDA通告2015年第91号)16. 关于发布医疗器械注册指定检验工作管理规定的通告(CFDA通告2015年第94号)17. 关于发布医疗器械工艺用水质量管理指南的通告(CFDA通告2016年第14号)18. 关于第三类医疗器械生产企业实施医疗器械生产质量管理规范有关事宜的通告(CFDA通告2016年第19号)19. 关于发布《医疗器械临床试验伦理审查申请与审批表范本》等六个文件的通告(CFDA通告2016年第58号)20. 关于发布医疗器械生产企业质量管理体系年度自查报告编写指南的通告(CFDA 通告2016年第76号)21. 关于发布第二批免于进行临床试验医疗器械目录的通告(CFDA通告2016年第133号)22. 关于发布2016年第二批医疗器械临床试验监督抽查项目的通告(CFDA通告2016年第143号)23. 总局关于发布医疗器械生产企业质量控制与成品放行指南的通告(CFDA通告2016年第173号)24. 总局关于发布医疗器械审评沟通交流管理办法(试行)的通告(CFDA通告2017年第19号)25.总局关于发布第三批免于进行临床试验医疗器械目录的通告(CFDA通告2017年第170号)26. 总局关于需审批的医疗器械临床试验申请沟通交流有关事项的通告(CFDA通告2017年第184号)27. 总局关于发布医疗器械注册单元划分指导原则的通告(CFDA通告2017年第187号)28. 总局关于发布移动医疗器械注册技术审查指导原则的通告(CFDA通告2017年第222号)29. 总局关于发布接受医疗器械境外临床试验数据技术指导原则的通告(CFDA通告2018年第13号)政策解读1. 医疗器械抽验和不良事件监测部分(一)(2015年01月22日发布)2. 《医疗器械经营监督管理办法》部分(一)(2015年01月22日发布)3. 《医疗器械生产监督管理办法》部分(一)(2015年01月22日发布)4. 医疗器械注册管理法规解读之一(《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》部分)(2015年02月05日发布)5. 医疗器械注册管理法规解读之二(《医疗器械说明书和标签管理规定》部分)(2015年02月05日发布)6. 关于《药品医疗器械飞行检查办法》的说明(2015年07月08日发布)7. 医疗器械注册管理法规解读之三(关于《医疗器械分类规则》的修订说明)(2015年07月16日)8. 医疗器械注册管理法规解读之四(2015年11月02日发布)9. 医疗器械注册管理法规解读之五(2015年11月19日发布)10. 医疗器械不良事件监测的主要目的和意义(2015年11月25日发布)11. 关于《医疗器械使用质量监督管理办法》的说明(2015年11月30日发布)12. 医疗器械注册管理法规解读之六(2016年01月07日发布)13. 关于《医疗器械通用名称命名规则》的说明(2016年01月27日发布)14. 《医疗器械临床试验质量管理规范》解读(2016年03月23日发布)15. 关于《医疗器械优先审批程序》的说明(2016年10月26日发布)16. 《医疗器械质量管理体系用于法规的要求》标准解读(一)(2017年02月04日发布)17. 《医疗器械质量管理体系用于法规的要求》标准解读(二)(2017年02月04日发布)18. 《医疗器械召回管理办法》解读(2017年02月08日发布)19. 《体外诊断试剂注册管理办法修正案》解读(2017年02月08日发布)20. 《医疗器械网络安全注册技术审查指导原则》解读(2017年03月02日发布)21. 《关于调整部分医疗器械行政审批事项审批程序的决定》解读(2017年04月06日发布)22. 《医疗器械标准管理办法》解读(2017年04月26日发布)23. 《医疗器械召回管理办法》解读之二(2017年05月24日发布)24. 图解政策:2017年器械注册管理改革有这4项重要举措(2017年07月14日发布)25. 图解政策:体外诊断试剂注册管理办法修正案(2017年07月28日发布)26. 创新医疗器械特别审批程序相关问题解读(2017年07月31日发布)27. 医疗器械临床试验质量管理相关问题解读(2017年07月31日发布)28. 《医疗器械标准管理办法》解读之一(2017年08月09日发布)29. 《医疗器械标准管理办法》解读之二(2017年08月09日发布)30. 《医疗器械网络安全注册技术审查指导原则》解读(2017年08月09日发布)31. 《医疗器械临床试验机构条件和备案管理办法》解读(2017年11月24日发布)32. 《医疗器械网络销售监督管理办法》解读(2017年12月22日发布)33. 《移动医疗器械注册技术审查指导原则》解读(2017年12月29日发布)通知性文件1. 关于进一步做好医疗器械产品分类界定工作的通知(食药监办械〔2013〕36号)2. 关于小型医用吸氧器监管有关问题的通知(食药监办法〔2013〕63号)3. 关于实施避孕套出口备案管理的通知(食药监办械监〔2013〕72号)4. 关于进一步加强医疗器械不良事件监测体系建设的指导意见(食药监械监〔2013〕205号)5. 关于印发医疗器械质量监督抽查检验管理规定的通知(食药监械监〔2013〕212号)6. 关于加强省级医疗器械技术审评能力建设的指导意见(食药监械管〔2013〕220号)7. 关于印发医疗器械生产日常监督现场检查工作指南的通知(食药监办械监〔2014〕7号)8. 关于印发创新医疗器械特别审批程序(试行)的通知(食药监械管〔2014〕13号)9. 关于认真贯彻实施《医疗器械监督管理条例》的通知(食药监法〔2014〕31号)10. 关于进一步做好医疗器械召回信息公开工作的通知(食药监办械监〔2014〕107号)11. 关于实施《医疗器械生产监督管理办法》和《医疗器械经营监督管理办法》有关事项的通知(食药监械监〔2014〕143号)12. 关于实施《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》有关事项的通知(食药监械管〔2014〕144号)13. 关于实施第一类医疗器械备案有关事项的通知(食药监办械管〔2014〕174号)14. 关于印发医疗器械检验机构开展医疗器械产品技术要求预评价工作规定的通知(食药监械管〔2014〕192号)15. 关于印发境内第三类和进口医疗器械注册审批操作规范的通知(食药监械管〔2014〕208号)16. 关于印发境内第二类医疗器械注册审批操作规范的通知(食药监械管〔2014〕209号)17. 关于印发医疗器械生产企业分类分级监督管理规定的通知(食药监械监〔2014〕234号)18. 关于印发国家重点监管医疗器械目录的通知(食药监械监〔2014〕235号)19. 关于启用医疗器械生产经营许可备案信息系统的通知(食药监办械监函〔2014〕476号)20. 关于加强避孕套质量安全管理的通知(食药监械监〔2015〕30号)21. 关于印发体外诊断试剂质量评估和综合治理工作方案的通知(食药监办械监〔2015〕55号)22. 关于印发境内第三类医疗器械注册质量管理体系核查工作程序(暂行)的通知(食药监械管〔2015〕63号)23. 关于对取消和下放行政审批事项加强事中事后监管的意见(食药监法〔2015〕65号)24. 关于印发医疗器械经营企业分类分级监督管理规定的通知(食药监械监〔2015〕158号)25. 关于印发医疗器械经营环节重点监管目录及现场检查重点内容的通知(食药监械监〔2015〕159号)26. 关于印发医疗器械生产质量管理规范现场检查指导原则等4个指导原则的通知(食药监械监〔2015〕218号)27. 关于印发医疗器械经营质量管理规范现场检查指导原则的通知(食药监械监〔2015〕239号)28. 关于执行医疗器械和体外诊断试剂注册管理办法有关问题的通知(食药监械管〔2015〕247号)29. 关于印发医疗器械检验机构资质认定条件的通知(食药监科〔2015〕249号)30. 关于成立医疗器械分类技术委员会的通知(食药监械管〔2015〕259号)31. 关于进一步加强医疗器械抽验工作的通知(食药监办械监〔2016〕9号)32. 关于切实做好第三类医疗器械生产企业实施医疗器械生产质量管理规范有关工作的通知(食药监办械监〔2016〕12号)33. 关于医疗器械产品技术要求有关问题的通知(食药监办械管〔2016〕22号)34. 总局关于实施《医疗器械通用名称命名规则》有关事项的通知(食药监械管〔2016〕35号)35. 总局关于印发一次性使用无菌注射器等25种医疗器械生产环节风险清单和检查要点的通知(食药监械监〔2016〕37号)。
附件
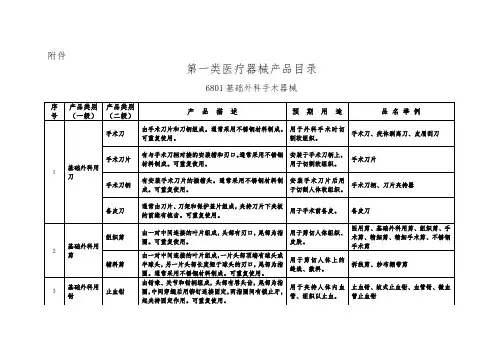
第一类医疗器械产品目录
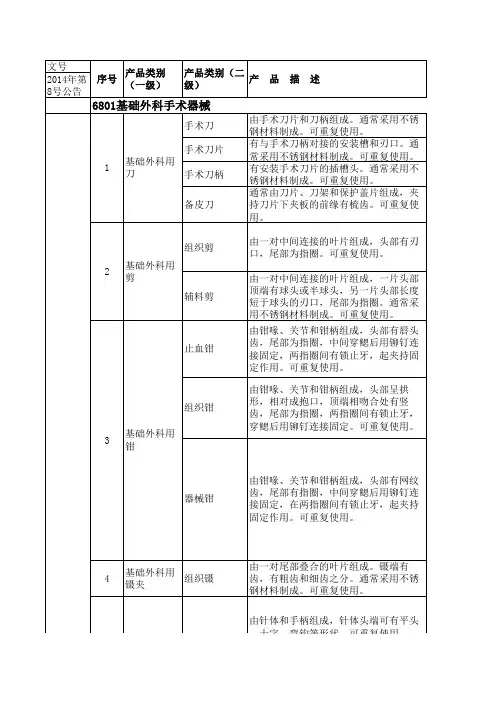
6801基础外科手术器械
6802显微外科手术器械
6803神经外科手术器械
6804眼科手术器械
6805耳鼻喉科手术器械
6806口腔科手术器械
6807胸腔心血管外科手术器械
6808腹部外科手术器械
6809泌尿肛肠外科手术器械
6810矫形外科(骨科)手术器械
6812妇产科用手术器械
6813计划生育手术器械
6815注射穿刺器械
6816烧伤(整形)科手术器械
6820普通诊察器械
6822医用光学器具、仪器及内窥镜设备
6823医用超声仪器及有关设备
6827中医器械
6831医用X射线附属设备及部件
6834医用射线防护用品、装置
6840临床检验分析仪器
6841医用化验和基础设备器具。
总局关于实施《医疗器械分类目录》有关事项的通告(2017年第143号)为贯彻落实《医疗器械监督管理条例》和《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号),国家食品药品监督管理总局于2017年8月31日发布《医疗器械分类目录》(以下简称新《分类目录》),自2018年8月1日起施行。
为做好新《分类目录》实施工作,现将有关事项通告如下:一、新《分类目录》的总体说明(一)新《分类目录》按技术专业和临床使用特点分为22个子目录,子目录由一级产品类别、二级产品类别、产品描述、预期用途、品名举例和管理类别组成。
判定产品的管理类别时,应当根据产品的实际情况,结合新《分类目录》中产品描述、预期用途和品名举例进行综合判定,产品描述和预期用途是用于判定产品的管理类别,不代表相关产品注册内容的完整表述。
注册申请人可以使用新《分类目录》的品名举例,或根据《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)拟定产品名称。
(二)新《分类目录》不包括体外诊断试剂,体外诊断试剂产品类别应当按照《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号,以下简称5号令)、《体外诊断试剂注册管理办法修正案》(总局令第30号,以下简称30号令)、《6840 体外诊断试剂分类子目录(2013版)》及后续发布的分类界定文件中有关体外诊断试剂的分类界定意见进行判定,分类编码继续延用6840。
(三)新《分类目录》不包括组合包类产品,组合包类产品的类别应当依据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、5号令、30号令等相关规定进行判定。
(四)《关于发布第一类医疗器械产品目录的通告》(国家食品药品监督管理总局通告2014年第8号)、《食品药品监管总局办公厅关于实施第一类医疗器械备案有关事项的通知》(食药监办械管〔2014〕174号)和2014年5月30日以后发布的医疗器械分类界定文件中有关第一类医疗器械产品的分类界定意见继续有效。
附件
第一类医疗器械产品目录
6801基础外科手术器械
6802显微外科手术器械
6803神经外科手术器械
6804眼科手术器械
6805耳鼻喉科手术器械
6806口腔科手术器械
6807胸腔心血管外科手术器械
6808腹部外科手术器械
6809泌尿肛肠外科手术器械
6810矫形外科(骨科)手术器械
6812妇产科用手术器械
6813计划生育手术器械
6815注射穿刺器械
6816烧伤(整形)科手术器械
6820普通诊察器械
6822医用光学器具、仪器及内窥镜设备
6823医用超声仪器及有关设备
6827中医器械
6831医用X射线附属设备及部件
6834医用射线防护用品、装置
6840临床检验分析仪器
6841医用化验和基础设备器具。
总局关于实施《医疗器械分类目录》有关事项的通告(2017年第143号)2017年09月04日发布为贯彻落实《医疗器械监督管理条例》和《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号),国家食品药品监督管理总局于2017年8月31日发布《医疗器械分类目录》(以下简称新《分类目录》),自2018年8月1日起施行。
为做好新《分类目录》实施工作,现将有关事项通告如下:一、新《分类目录》的总体说明(一)新《分类目录》按技术专业和临床使用特点分为22个子目录,子目录由一级产品类别、二级产品类别、产品描述、预期用途、品名举例和管理类别组成。
判定产品的管理类别时,应当根据产品的实际情况,结合新《分类目录》中产品描述、预期用途和品名举例进行综合判定,产品描述和预期用途是用于判定产品的管理类别,不代表相关产品注册内容的完整表述。
注册申请人可以使用新《分类目录》的品名举例,或根据《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)拟定产品名称。
(二)新《分类目录》不包括体外诊断试剂,体外诊断试剂产品类别应当按照《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号,以下简称5号令)、《体外诊断试剂注册管理办法修正案》(总局令第30号,以下简称30号令)、《6840 体外诊断试剂分类子目录(2013版)》及后续发布的分类界定文件中有关体外诊断试剂的分类界定意见进行判定,分类编码继续延用6840。
(三)新《分类目录》不包括组合包类产品,组合包类产品的类别应当依据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、5号令、30号令等相关规定进行判定。
(四)《关于发布第一类医疗器械产品目录的通告》(国家食品药品监督管理总局通告2014年第8号)、《食品药品监管总局办公厅关于实施第一类医疗器械备案有关事项的通知》(食药监办械管〔2014〕174号)和2014年5月30日以后发布的医疗器械分类界定文件中有关第一类医疗器械产品的分类界定意见继续有效。
国家食品药品监督管理总局
通告
2014年第13号
关于发布免于进行临床试验的第三类医疗器械目录的通告
为了做好医疗器械注册管理工作,根据《医疗器械监督管理条例》(国务院令第650号)和《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号),国家食品药品监督管理总局组织制定了《免于进行临床试验的第三类医疗器械目录》,现予发布,自2014年10月1日起施行。
特此通告。
附件:免于进行临床试验的第三类医疗器械目录
国家食品药品监督管理总局
2014年8月21日
2014年第13号通告附件.doc
附件
免于进行临床试验的第三类医疗器械目录
国家食品药品监督管理总局
通告
2014年第12号
关于发布免于进行临床试验的第二类医疗器械目录的通告
为做好医疗器械注册管理工作,根据《医疗器械监督管理条例》(国务院令第650号)和《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号),国家食品药品监督管理总局组织制定了《免于进行临床试验的第二类医疗器械目录》,现予发布,自2014年10月1日起施行。
特此通告。
附件:免于进行临床试验的第二类医疗器械目录
国家食品药品监督管理总局
2014年8月21日
2014年第12号通告附件.doc
通告2014年第12号附件
免于进行临床试验的第二类医疗器械目录
附件
免于进行临床试验的第二类医疗器械目录。
国家食品药品监督管理总局令第8 号《医疗器械经营监督管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。
局长张勇2014年7月30日医疗器械经营监督管理办法第一章总则第一条为加强医疗器械经营监督管理,规范医疗器械经营行为,保证医疗器械安全、有效,根据《医疗器械监督管理条例》,制定本办法。
第二条在中华人民共和国境内从事医疗器械经营活动及其监督管理,应当遵守本办法。
第三条国家食品药品监督管理总局负责全国医疗器械经营监督管理工作。
县级以上食品药品监督管理部门负责本行政区域的医疗器械经营监督管理工作。
上级食品药品监督管理部门负责指导和监督下级食品药品监督管理部门开展医疗器械经营监督管理工作。
第四条按照医疗器械风险程度,医疗器械经营实施分类管理。
经营第一类医疗器械不需许可和备案,经营第二类医疗器械实行备案管理,经营第三类医疗器械实行许可管理。
第五条国家食品药品监督管理总局制定医疗器械经营质量管理规范并监督实施。
第六条食品药品监督管理部门依法及时公布医疗器械经营许可和备案信息。
申请人可以查询审批进度和审批结果,公众可以查阅审批结果。
第二章经营许可与备案管理第七条从事医疗器械经营,应当具备以下条件:(一)具有与经营范围和经营规模相适应的质量管理机构或者质量管理人员,质量管理人员应当具有国家认可的相关专业学历或者职称;(二)具有与经营范围和经营规模相适应的经营、贮存场所;(三)具有与经营范围和经营规模相适应的贮存条件,全部委托其他医疗器械经营企业贮存的可以不设立库房;(四)具有与经营的医疗器械相适应的质量管理制度;(五)具备与经营的医疗器械相适应的专业指导、技术培训和售后服务的能力,或者约定由相关机构提供技术支持。
从事第三类医疗器械经营的企业还应当具有符合医疗器械经营质量管理要求的计算机信息管理系统,保证经营的产品可追溯。
鼓励从事第一类、第二类医疗器械经营的企业建立符合医疗器械经营质量管理要求的计算机信息管理系统。