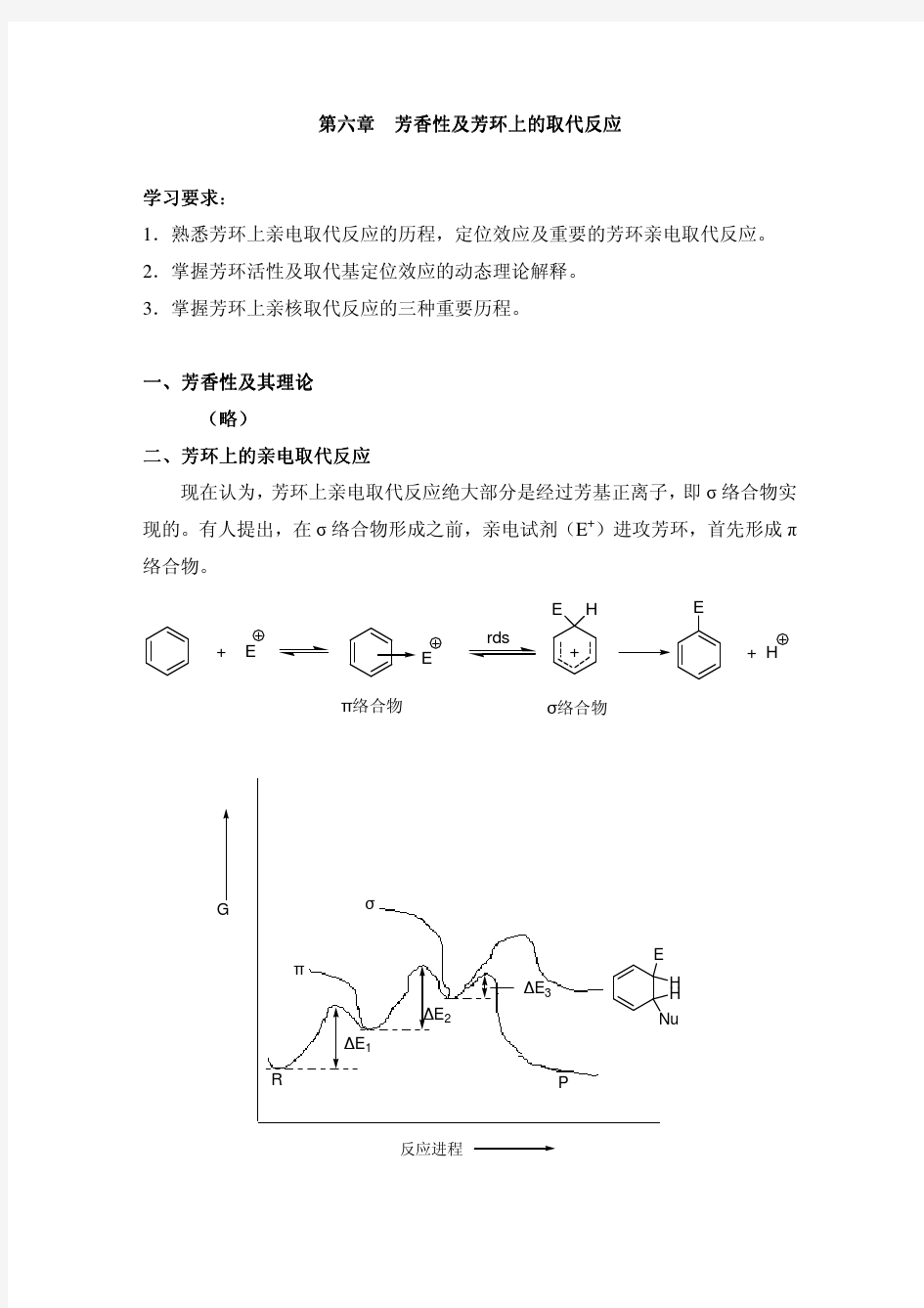

芳环上的取代氯化反应 1 反应历程 1.1以金属卤化物为催化剂:FeCl3,AlCl3,ZnCl2 1.2以硫酸为催化剂 1.3以碘为催化剂 1.4以次卤酸为催化剂 苯环上有吸电子取代基,反应较难进行,需要加入催化剂;苯环上有供电子取代基,反应容易进行,可以不用催化剂。 2 反应动力学及氯化深度
表 苯在氯化、磺化和硝化反应中k 1/k 2的比较 表 氯苯含量对k 1/k 2的影响 氯化液中氯苯浓度与反应液中苯浓度的关系式: K 与反应温度、搅拌效果有关。 氯化液中氯苯浓度的极大值: 此时氯化液中苯的浓度: 图 苯在间歇氯化时的产物组成变化 每摩尔纯苯所消耗的氯气的量(摩尔)叫做氯化深度。 3 芳环取代氯化反应的影响因素 3.1原料纯度 1)水份:<0.04% 2)氢气含量:<4% 3)噻吩:使催化剂中毒;腐蚀设备 4)循环使用的苯(苯氯比4:1)中氯苯的影响 3.2氯化深度 3.3混合作用 在连续反应时,由于反应器型式选择不当、传质不均匀,使反应生成的产物未能及时离开,又返回反应区域促进连串反应的进行,这种现象叫做反混作用。 C C C C
(a)单锅间歇生产工艺 (b)多锅连续生产工艺图 (c)塔式沸腾连续生产工艺 图 氯苯的生产工艺 3.4氯化温度 表 苯氯化反应温度与k 2/k 1的关系 3.5 催化剂 3.6反应介质 液态:无需溶剂 固态:水 浓硫酸、发烟硫酸、氯磺酸等 有机溶剂 4 芳环取代氯化反应实例 4.1氯苯的生产 1)直接氯化法 Cl 2 FeCl 3 ℃O 22H Cl +NaC lO O 22,H 2O 浓H 2 S O 4 ,90~100 ℃ C l ,I ,催化 C l ,110℃ 乙酸或氯苄介质
芳香族亲电取代反应的研究 摘要:总结了芳香族亲电取代反应的过程,并对其反应机理进行深入的探讨,对典型的 芳香族亲电取代反应进行了归纳,同时讨论了芳香族化合物的结构和性能的关系。 关键词:亲电取代;反应机理 1芳香族亲电取代反应总论 苯的芳香性结构决定了苯的稳定性。苯环上的:电子为芳香取代反应供给了电子,很多亲电试剂能与之反应,其范围远比烯、炔加成反应广泛。可以用一个统一的反应机理来解释这些取代反应,虽然反应的动力学、自由能—反应曲线对每一种亲电试剂来说可以是不一样的,但若干中间步骤基本上是相似的,这就允许用一个亲电试剂E+做为总的代表反应的一个组成部分,芳环的二体系也参加到反应中去,可以中间体或过渡态的形式出现,有时是一种Π络合物,但它很快转变为。δ络合物,后者以环上的某一个碳与亲电试剂结合,这个碳也就是取代反应发生的位置。δ和Π二络合物的形成和解体是可逆的,从络合物上退减后才得到稳定的芳香体系,究竟退减是H还是E,要看哪一个易于被排除,对大多数取代反应来说,退减一个质子容易的话,这一取代反应是不可逆的,但在某些条件下,如果亲电取代基团是可以被H所排除的,这个反应就是可逆的。δ络合物是反应活性和定位的关键。硝化反应作为不可逆亲电取代反应的代表,已经证明过亲电试剂是硝基正离子NO2十,从硝酸离解出NO2十是决定性的步骤。动力学研究发现有两种速度规律,对于相对不活泼的芳香底物,速度是二级的,硝化试剂一级,芳香化合物也是一级,这相当于亲电试剂和芳香底物的相互结合成为速度最慢步骤.对比较活泼的芳香底物,这一步快于硝基正离子的生成速度,于是速度式中没有芳香化合物浓度一项,在这种情况下,各类芳香性化合物的硝化反应速度相等,都取决于硝酸浓度一项。通常在芳香取代反应中亲电试剂的活性是很大的,所得中间体也很活泼,生成亲电试剂的过程可能是决定反应速度的一步,但也不一定具有决定性的作用。 1.1、反应机理 1.芳环亲电取代反应的一般机理 1.2下列为常见的亲电试剂及其发生的形式:
苯环上亲电取代反应的定位规律 基本概念:定位基:在进行亲电取代反应时,苯环上原有取代基,不仅影响着苯环的取代反应活性,同时决定着第二个取代基进入苯环的位置,即决定取代反应的位置。原有取代基称做定位基。 一、两类定位基 在一元取代苯的亲电取代反应中,新进入的取代基可以取代定位基的邻、间、对位上的氢原子,生成三种异构体。如果定位基没有影响,生成的产物是三种异构体的混合物,其中邻位取代物40%(2/5)、间位取代物40%(2/5)和对位取代物20%(1/5)。实际上只有一种或二种主要产物。例如各种一元取代苯进行硝化反应,得到下表所示的结果: 排在苯前面的取代硝化产物主要是邻位和对位取代物,除卤苯外,其它取代苯硝化速率都比苯快;排在苯后面取代硝化产物主要是间位取代物,硝化速率比苯慢得多。归纳大量实验结果,根据苯环上的取代基(定位基)在亲电取代反应中的定位作用,一般分为两类:第一类定位基又称邻对位定位基:—O-,—N(CH3)2,—NH2,—OH,—OCH3,—NHCOCH3,—OCOCH3,—F,—Cl,—Br,—I,—R,—C6H5等。 第二类定位基又称间位定位基:—N+(CH3)3,—NO2,—CN,—SO3H,—CHO,—COCH3,—COOH,—COOCH3,—CONH2,—N+H3等。 两类定位基的结构特征:第一类定位基与苯环直接相连的原子上只有单键,且多数有孤对电子或是负离子;第二类定位基与苯环直接相连的原子上有重键,且重键的另一端是电负性
大的元素或带正电荷。两类定位基中每个取代基的定位能力不同,其强度次序近似如上列顺 序。 苯环上亲电取代反应的定位规律 二、定位规律的电子理论解释 在一取代苯中,由于取代基的电子效应沿着苯环共轭链传递,在环上出现了电子云密度较 大和较小的交替分布现象,因而环上各位置进行亲电取代反应的难易程度不同,出现两种定 位作用。也可以从一取代苯进行亲电取代反 应生成的中间体σ络合物的相对稳定性的角度进行考察,当亲电试剂E +进攻一取代 时,生成三苯 σ络合物: Z 不同,生成的三种σ 络合物碳正离子的稳定性不同,出现了两种定位作用。 1.第一类定位基对苯环的影响及其定位效应 以甲基、氨基和卤素原子为例说明。 甲基在甲苯中,甲基的碳为sp3杂化,苯环碳为sp2杂化,sp2杂化碳的电负性比sp3杂 化碳的大,因此,甲基表现出供电子的诱导效应(A)。另外,甲基C—H σ 键的轨道与苯 环的π 轨道形成σ—π 超共轭体系(B)。供电诱导效应和超共轭效应的结果,苯环上电 子密度增加,尤其邻、对位增加得更多。因此,甲苯进行亲电取代反应比苯容易,而且主要 发生在邻、对位上。 亲电试剂E+进攻甲基的邻、间、对位置,形成三种σ 络合物中间体,三种σ 络合物 碳正离子的稳定性可用共振杂化体表示: 进攻邻位:
亲和取代反应总结
亲核取代反应总结 1、反应定义: 亲核取代反应(Nucleophilic Substitution Reaction)是指有机分子中与碳相连的某原子或基团被作为亲核试剂的某原子或基团取代的反应。在反应过程中,取代基团提供形成新键的一对电子,而被取代的基团则带着旧键的一对电子离去。 2、反应意义: 这类反应是有机化学中非常重要的一类反应,不论在理论研究中还是在有机合成实际中都是极其有用的一类反应。 3、反应分类: 亲核取代反应的主要类型为脂肪族饱和碳上的亲核取代反应,即饱和卤代烃与亲核试剂的取代反应,较特殊结构的有苄基卤代物、烯丙基卤代物亲核反应。其他类型还包括与酰氯、磺酸酯、磺酰卤、卤代苯等的取代反应。 从电荷类型来分,亲核取代反应只能有四种类型: (1)中性底物和负离子亲核试剂反应 (2)中性底物和中性亲核试剂反应 (3)正离子底物和负离子亲核试剂反 (4) 正离子底物和中性亲核试剂反应
亲核试剂包括有机和无机两类分子或离子: 无机类亲核试剂:OH-、CN-、X-、H2O、NH3等 有机类亲核试剂:ROH、RO-、PhO-、RS-、RMgX、RCOO-等4、反应机理类型分类: (1)双分子亲核取代反应(S N2) 有两种分子参与了决定反应速率关键步骤的亲核取代反应称为双分子亲核取代反应。反应过程中,亲核试剂从反应物离去基团的背面向与它连接的碳原子进攻,先与碳原子形成比较弱的键,同时离去基团与碳原子的键有一定程度的减弱,两者与碳原子成一条直线,碳原子上另外三个键逐渐由伞形转变成平面,这需要消耗能量,即活化能,当反应进行和达到能量最高状态即过渡态后,亲核试剂与碳原子之间的键开始形成,碳原子与离去基团之间的键断裂,碳原子上三个键由平面向另一边偏转,整个过程犹如大风将雨伞由里向外反转一样,这时就要释放能量,形成产物,S N2反应机理一般式表示为: Nu-+R X [Nuδ-···R···Xδ- ] NuR+X- 例如,溴甲烷与OH-的水解反应:
第七章 芳环上的亲电和亲核取代反应 7.1芳环的亲电取代反应 7.1.1芳环上的亲电取代历程 1、 亲电试剂的产生 HNO 3+2H 2SO 4 NO 2++H 3O ++2HSO 4- 亲电试剂 2、 π-络合物的形成 芳环上电子云密度 R +NO 2 π -络合物NO 2 3、 σ-络合物的形成 NO 2+ H NO 2 σ-络合物 硝基所在碳为sp 3 杂化 4、 消去-H + + NO 2 H NO 2 快 σ-络合物的证据 已分离出C + CH 3 +CH 3CH 2F +BF 3 CH 3 CH 2CH 3 H BF 4- 通过红外和核磁等可鉴定中间体的结构。
CH 3 CH 3 CH 3EtF 3CH 3 H Et CH CH 3 BF 4- 3 mp - 15 C 7.1.2苯环上亲电取代反应的定位规律 从反应速度和取代基进入的位置进行考虑 1、 第一类定位基(邻,对位定位基) 致活基 NH 2 NR 2 OH OR NHCR O Ph R 致钝基 F Cl Br I 2、 第二类定位基(间位定位基) 均为致钝基 NO 2 NR 3 COOH COOR SO 3H CN CHO CR O CCl 3 7.1.3定位规律在有机合成中的应用 7.1.4典型的芳香亲电取代反应 1.硝化反应 硝化试剂有HNO 3-H 2SO 4 HNO 3+2H 2SO 4 NO 2++H 3O ++2HSO 4- 真正的硝化试剂为硝酰正离子。混酸体系有强氧化性 OH 20%HNO 3 OH NO 2 + OH 2 如用混酸将得氧化产物 NH 2 浓HNO 浓H 2SO 4 NH 3HSO 4- NO 2 同时还有部分氧化产物 HNO 3/CCl 4低温时的硝化速度较快 (HNO 3)xH 2NO 3+ NO 2++H 2O +(HNO 3)x 0℃
第七章芳环上的亲电和亲核取代反应7.1芳环的亲电取代反应 7.1.1芳环上的亲电取代历程 1、亲电试剂的产生 亲电试剂 2、π-络合物的形成 芳环上电子云密度R 3、σ-络合物的形成 硝基所在碳为sp3杂化 4、消去-H+ σ-络合物的证据 已分离出C+ 通过红外和核磁等可鉴定中间体的结构。
7.1.2苯环上亲电取代反应的定位规律 从反应速度和取代基进入的位置进行考虑 1、第一类定位基(邻,对位定位基) 2、第二类定位基(间位定位基) 均为致钝基 7.1.3定位规律在有机合成中的应用 7.1.4典型的芳香亲电取代反应 1.硝化反应 硝化试剂有HNO3-H2SO4 真正的硝化试剂为硝酰正离子。混酸体系有强氧化性 如用混酸将得氧化产物 同时还有部分氧化产物HNO3/CCl4低温时的硝化速度较快 温和的硝化试剂HNO2/C(NO2)4
可避免间位硝化与氧化 2.磺化反应 亲电试剂为SO3或(共轭酸) 特点:(1)可逆反应,可用于芳环的定向取代(占位)。(2)置换反应,合成一些难于合成的物质。 发生间接硝化 3.卤化反应 (1)卤素作卤化剂
(2)N-卤代酰胺或N-卤代磺酰胺作卤化剂 等 其卤化性能较差,只与活泼芳烃反应,可避免氧化反应发生(芳胺和酚)。 而在非极性CCl4等溶剂中是自由基引发剂 自由基取代反应。 1. Fridel-Crafts反应 (1) 烃基化 亲电试剂产生 催化剂活性 AlCl3>FeCl3>SbCl5>SnCl4>BF3>TiCl4>ZnCl2 特点 A. 易发生重排反应
亲电试剂发生重排 B. 易发生多烷基化 C. 可逆反应 动力学条件下,遵守定位规律,热力学控制条件下得稳定的间位产物。 D. 钝化的芳烃不发生烷基化 E.-OH,-NH2和-OR等与路易斯酸配位,使催化剂难于发生烷基化,可改用烯作烷基化剂,以酚铝或苯胺铝作催化剂 (2)酰基化反应 1 用酰氯时,ACl3的量要大于1mol,用酸酐时ACl3要大于2mol。 2 酚的酰化是Fries重排 3 不会发生重排 5.重氮盐的偶联反应
.磺化反应特点及副反应 1.磺化反应是一种平衡的可逆反应 磺化是典型的亲电取代反应,以硫酸为磺化剂,以最常见的芳环的磺化为例,反应过程一般认为如下: 从上式可以看出,是正离子对芳环进行进攻,正离子的浓度和体系中的含水量有重要的关系,因为反应是平衡反应,水量越少正离子越多。 2.磺酸基容易被水解 芳磺酸在含水的酸性介质中,会发生水解使磺基脱落,是硫酸磺化历程的逆反应。 对于有吸电子基的芳磺酸,芳环上的电子云密度降低、磺基难水解。对于有给电子的芳磺酸,芳环上的电子云密度较高,磺基容易水解。介质中酸离子浓度愈高,水解速度愈快,因此磺酸的水解采用中等浓度的硫酸。磺化和水解的反应速度都与温度有关,温度升高时,水解速度增加值比磺化速度快,因此一般水解的温度比磺化的温度高。
3.磺酸的异构化 磺化时发现,在一定条件下,磺基会从原来的位置转移到其它的位置,这种现象称为“磺酸的异构化”,在有水的硫酸中,磺酸的异构化是一个水解再磺化的过程,而在无水溶液中则是分子内的重排过程。 温度的变化对磺酸的异构化也有一定的影响,在苯、萘及其衍生物中将有论述。 4.副反应 磺化反应最主要的副反应是形成砜,特别是在芳环过剩和磺化剂活性强(如发烟硫酸、三氧化硫、氯磺酸)的时候。如: 用三氧化硫磺化时,极易形成砜,可以用卤代烷烃为溶剂,也可以用三氧化硫和二氧六环、吡啶等的复合物来调节三氧化硫的活性。发烟硫酸磺化时,可通过加入无水硫酸钠,抑制砜的形成。无水硫酸钠在萘酚磺化时,可抑制硫酸的氧化作用。
四.磺化反应的影响因素 1.被磺化物的结构 磺化反应是典型的亲电取代反应,当芳环上有给电子基团时,磺化反应较易进行,如-Cl、-CH2、–OH、–NH2,磺基进入该类取代基的对位。磺基所占的空间较大,所以邻位的产品较少,特别是当芳环上的取代基所占空间较大时尤为明显。从表7-2-01可以看出,在烷基苯磺化时,邻位磺酸的生成量随烷基的增大而减少。 芳环上有吸电子基团时,对磺化反应不利。如硝基、羧基的存在,使其磺化的速度较苯环降低。 2.磺化剂的浓度和用量 磺化动力学研究指出,硫酸浓度稍有变化对磺化速度就有显著影响。在92~99%浓硫酸中,磺化速度与硫酸中所含水分浓度平方成反比。采用硫酸作磺化剂时,生成的水将使进一步磺化的反应速度大为减慢。当硫酸浓度降至某一程度时,反应即自行停止。此时剩余的硫酸叫做废酸,习惯把这种废酸以三氧化硫的重量百分数表示,称之为“Л值”。显然,对于容易磺化的过程,Л值要求较低。而对于难磺化的过程,Л值要求较高。有时废酸的浓度高于100%硫酸,即Л值大于81.6,各种芳烃的Л值见表7-2-02。
芳环上的取代磺化 一、磺化反应的历程及动力学 磺化反应:一种向有机分子中引入磺酸基(—SO3H)或磺酰氯基(—SO3Cl)的反应过程,磺化是磺基(或磺酰卤基)中的硫原子与有机分子中的碳原子相连接形成C-S键的反应,生成的产物是磺酸(R—SO3H,R表示烃基)、磺酸盐(R—SO3M;M表示NH4或金属离子)或磺酰氯(R—SO2Cl)。在这儿着重介绍芳环上的取代磺化。 磺化是亲电取代反应,因此芳环上有供电基使磺化反应速率变快,有吸电基使磺化反应速率变慢。磺化也是连串反应,但是与氯化不同,磺酸基对芳环有较强的钝化作用,一磺酸比相应的被磺化物难于磺化,而二磺酸又比相应的一磺酸难于磺化。因此,苯系和萘系化合物在磺化时,只要选择合适的反应条件,例如磺化剂的浓度和用量、反应的温度和时间,在一磺化时可以使被磺化物基本上完全一磺化,只副产很少量的二磺酸;在二磺化时只副产很少量的三磺酸。例如,在苯的共沸去水一磺化时,磺化液中约含有88%~91%苯磺酸、小于 15%苯、小于0.5%苯二磺酸和2.0%~4%硫酸(均为质量分数,下同)。 芳香化合物进行磺化反应时,分两步进行。首先,亲电质点向芳环进行亲电进攻,生成σ-配合物,后在碱作用下脱去质子得到芳环酸。反应历程如下: 研究证明,用浓硫酸磺化时,脱质子较慢,第二步是整个反应速率的控制步骤。用稀酸磺化时,生成σ-配合物较慢,第一步限制了整个反应速率。 采用发烟硫酸或硫酸磺化芳烃时,其反应动力学可如下表示。 当磺化质点为SO3 时:v=kso3[ArH][SO3]= k′SO3[ArH][H2O]2- 当磺化质点为H2S2O7时:v=kSA[ArH][H2S2O7]=k′H2S2O7[ArH][H2O]2-当磺化质点为H3SO4 +时:v=kH3SO4 +[ArH][H3SO4 +]=k′H3SO4 +[ArH][H2O]- 由以上三式可以看出,磺化反应速率与磺化剂中的含水量有关。当以浓硫酸为磺化剂,水很少时,磺化反应速率与水浓度的平方成反比,即生成的水量越多,反应速率下降越快。因此,用硫酸作磺化剂的磺化反应中,硫酸浓度及反应中生成的水量多少对磺化反应速率的影响是一个十分重要的因素。 二、磺化目的: 1) 赋予有机物酸性、水溶性、表面活性及对纤维的亲和力等。 2) 可将-SO3H转化为其它基团。 3)利用-SO3H可水解性,辅助定位或提高反应活性。
亲核取代反应 一.亲核取代反应机理。 亲核取代反应是指有机分子中的与碳相连的原子或原子团被作为亲核试剂的某原子或原子团取代的反应。反应分为SN1型(单分子取代反应),与SN2型双分子取代反应。 1.SN1型(单分子取代反应) 第一步是碳原子上正电荷增加,离去基团负点性增加,经过过渡态(1)并最终解离,生成活性中间体碳正离子与离去基团负离子。由于这一步反应的活化能较高,速率较慢,所以这一步是反应的决速步。 第二步是活性中间体的碳正离子与亲和试剂作用,生成反应产物。这一步仅需少量能量,速率很快。 反应特点: (1)SN1反应的决速步是中心碳原子与离去基团之间化学键的异裂。反应速率只取决 于一种分子的浓度,因此,它在动力学上是一级反应。 (2)一般是一个两步反应。第一步生成的碳正离子采取SP2杂化,是平面构型。故 若反应物的中心碳原子是手性碳,反应产物一般是一对等量的对映异构体的混 合物——外消旋体。 (3)反应中间体生成的碳正离子导致反应有重排的趋势。 2.SN2型(双分子取代反应) 反应中,离去基团离开中心碳原子的同时,亲核试剂与中心碳原子发生部分键合,无中间体生成。 有机反应中,将两种分子参与决速步的亲核取代反应陈伟双分子亲核取代反应。 反应特点: (1)SN2反应是一步反应,只有一个过渡态。 (2)在SN2反应中,亲核试剂进攻中心碳原子是总是从离去基团溴原子的背面沿着 碳原子和离去基团连接的中心线方向进攻。这个过程会使得碳原子与三个未参 与反映的键发生翻转,这种翻转称为瓦尔登翻转,又称构型翻转。 二.影响亲核取代反应的因素 1.烃基结构的影响。 对SN1反应,主要考虑碳正离子的稳定性。对SN2反应,主要取决于过渡态形 成的难易,也就是空间效应的影响。 2.离去基团的影响。
芳环上的取代氯化反应机理 1 反应历程 1.1以金属卤化物为催化剂:FeCl3,AlCl3,ZnCl2 1.2以硫酸为催化剂 1.3以碘为催化剂 1.4以次卤酸为催化剂 苯环上有吸电子取代基,反应较难进行,需要加入催化剂;苯环上有供电子取代基,反应容易进行,可以不用催化剂。 2 反应动力学及氯化深度
表 苯在氯化、磺化和硝化反应中k 1/k 2的比较 反应类型 氯化 磺化 硝化 k 1/k 2 8.5 103~104 105~107 表 氯苯含量对k 1/k 2的影响 氯苯含量,% 1 73.5 k 1/k 2 842 约1 氯化液中氯苯浓度与反应液中苯浓度的关系式: K 与反应温度、搅拌效果有关。 氯化液中氯苯浓度的极大值: 此时氯化液中苯的浓度: 图 苯在间歇氯化时的产物组成变化 每摩尔纯苯所消耗的氯气的量(摩尔)叫做氯化深度。 3 芳环取代氯化反应的影响因素 3.1原料纯度 1)水份:<0.04% 2)氢气含量:<4% 3)噻吩:使催化剂中毒;腐蚀设备 4)循环使用的苯(苯氯比4:1)中氯苯的影响 3.2氯化深度 3.3混合作用 在连续反应时,由于反应器型式选择不当、传质不均匀,使反应生成的产物未能及时离开,又返回反应区域促进连串反应的进行,这种现象叫做反混作用。 S Cl Cl Cl Cl Cl S Cl Cl Cl Cl
(a)单锅间歇生产工艺 (b)多锅连续生产工艺图 (c)塔式沸腾连续生产工艺 图 氯苯的生产工艺 3.4氯化温度 表 苯氯化反应温度与k 2/k 1的关系 T ,℃ 18 25 30 k 2/k 1 0.107 0.118 0.123 3.5催化剂 3.6反应介质 液态:无需溶剂 固态:水 浓硫酸、发烟硫酸、氯磺酸等 有机溶剂 4 芳环取代氯化反应实例 4.1氯苯的生产 1)直接氯化法 Cl 2FeCl 3 O 2N 2HCl+NaClO O 2N NH 2Cl 2浓H 2SO 4,90~100℃ O O Cl Cl Cl 2,I 2,催化 OH COOH OH COOH 2Cl
排在苯前面的取代硝化产物主要是邻位和对位取代物,除卤苯外,其它取代苯硝 化速率都比苯快;排在苯后面取代硝化产物主要是间位取代物,硝化速率比苯慢得多。归纳 大量实验结果,根据苯环上的取代基(定位基)在亲电取代反应中的定位作用,一般分为两 类: 第一类定位基又称邻对位定位基:—O-,—N(CH3)2,—NH2,—OH,—OCH3,—NHCOCH3, —OCOCH3,—F,—Cl,—Br,—I,—R,—C6H5等。 第二类定位基又称间位定位基:—N+(CH3)3,—NO2,—CN,—SO3H,—CHO,—COCH3,—COOH,—COOCH3,—CONH2,—N+H3等。 两类定位基的结构特征:第一类定位基与苯环直接相连的原子上只有单键,且多数 有孤对电子或是负离子;第二类定位基与苯环直接相连的原子上有重键,且重键的另一端是 电负性大的元素或带正电荷。两类定位基中每个取代基的定位能力不同,其强度次序近似如 上列顺序。 苯环上亲电取代反应的定位规律 二、定位规律的电子理论解释 在一取代苯中,由于取代基的电子效应沿着苯环共轭链传递,在环上出现了电子云 密度较大和较小的交替分布现象,因而环上各位置进行亲电取代反应的难易程度不同,出现 两种定位作用。也可以从一取代苯进行亲电取代反 应生成的中间体σ络合物的相对稳定性的角度进行考察,当亲电试剂 E+进攻一取代苯时,生成三σ络合物: Z 不同,生成的三种σ 络合物碳正离子的稳定性不同,出现了两种定位作用。 1.第一类定位基对苯环的影响及其定位效应 以甲基、氨基和卤素原子为例说明。 甲基在甲苯中,甲基的碳为 sp3杂化,苯环碳为 sp2杂化,sp2杂化碳的电负性 比 sp3杂化碳的大,因此,甲基表现出供电子的诱导效应(A)。另外,甲基 C—H σ 键 的轨道与苯环的π 轨道形成σ—π 超共轭体系(B)。供电诱导效应和超共轭效应的结 果,苯环上电子密度增加,尤其邻、对位增加得更多。因此,甲苯进行亲电取代反应比苯容 易,而且主要发生在邻、对位上。 亲电试剂 E+进攻甲基的邻、间、对位置,形成三种σ 络合物中间体,三种σ 络 合物碳正离子的稳定性可用共振杂化体表示:
亲电取代反应 亲电取代反应是亲电试剂进攻化合物负电部分,取代其它基团的化学反应。一般发生于芳香族化合物,是一种向芳香环系引入官能团的重要方法,是芳香族化合物的特性之一。被取代的基团通常是氢原子,但其他基团被取代的情形也是存在的。一般来说,亲电取代特指芳香亲电取代。另一种比较少见的亲电取代反应是脂肪族的亲电取代。 中文名 亲电取代反应 外文名 Electrophilic Substitution 属性 亲电取代 性质 反应 主要反应 硝化反应,卤化反应磺化反应等。 目录 .1原理
.2主要反应 .?硝化反应 .?卤化反应 .?磺化反应 .3定位规则 原理 亲电取代反应主要发生在芳香体系或富电子的不饱和碳上,就本质而言均是较强亲电基团对负电子体系进攻,取代较弱亲电基团。但对于芳香体系和脂肪体系,由于具体环境不同,其反应历程亦有所不同,现分述如下。 亲电芳香取代反应(electrophilic aromatic substitution)是芳香体系最重要的有机反应之一,常用于向芳香环系引入官能团,因此研究时间较长,在机理方面已基本达成一致。 主要反应 对于亲电取代反应,其最为主要的反应类型均在芳香体系中产生,所以这里仅仅对芳香亲电取代进行一定的举例介绍。 硝化反应 硝化反应苯环体系一个重要的反应,其常用于向体系引入硝基或利用硝基引入氨基等其他各种官能团,有很强的泛用性,定位选择性较好,使用最多。由于硝基有较强氧化性,而有机体系本身又具有一定的还原性,硝基含量较多的体系就很容易成为良好的炸药材料,其中著名的TNT、苦味酸等就是通过硝化反应制备的。 Friedel(傅瑞德尔)-Crafts(克拉夫茨)反应