
收稿日期:2012-06-11
作者简介:曾培龙(1987-),男,河南商丘人,硕士研究生,主要研究方向:生物信息学;
王亚东(1964-),男,辽宁锦州人,硕士,教授,博士生导师,主要研究方向:人工智能、机器学习、知识工程等。
0引言
新一代测序技术正在引领生命科学研究进入一个崭新阶段。人类基因组计划完成之后,获得个体基因组的全部序列对于生物学研究、探索与认识生命的本质具有十分重要的科学意义[1,2]。
新一代测序技术作为目前生命科学研究的基础手段,随着应用领域的迅速扩增与不断深入,对生物信息学提出了必须正视的基础研究课题。而全基因组序列拼接作为生物信息学的核心问题,面临的主要挑战有:
(1)海量的数据(覆盖深度一般为40-200倍,数据量达20-200GB ),
迫切需要海量数据的拼接组装算法;(2)测序数据中的错误,容易导致错拼;
(3)基因组中重复片段大量存在,
由于读取片段reads 长度过短,一般只有几十个碱基,这使得重复序列的处理变得困难。
针对新一代测序数据reads 长度较短、数据海量的特点,全基因组测序方面的数据分析软件的研发,已成为生物信息学领域最迫切、最重要的研究课题。虽然目前已开发有一些全基因组拼接软件,但是基本都局限在大型计算平台上完成数据分析过程,难以满足一般的研究需求,而且数据处理速度仍然远远落后于数据产生速度,已经成为整个基因组图谱绘制工作的瓶颈,并且其拼接结果在准确性方面还有待提高。
1全基因组序列拼接的含义
基因组序列拼接的核心思想是利用序列之间的交叠关
系,通过类似于“搭积木”的方式重建目标基因组序列。其
基本方法是将序列之间的交叠关系转换成计算机可以识别的结构,通过不断迭代扩展的方式延长目标序列,然后利用配对数据,确定各个目标序列的相对方向和位置关系,最终还原目标基因组序列。
基于新一代测序数据的基因组序列拼接,通常分为如下三个阶段:
(1)数据的预处理阶段。该阶段通过特定的方法,移除测序数据中的错误碱基;
(2)基因组连续片段(contigs )生成阶段。该阶段将reads 拼接成contigs ;
(3)超长序列片段(scaffoldings )组装阶段。该阶段使用配对数据,确定contigs 之间的方向和位置关系,生成scaffoldings 。
2全基因组序列拼接的发展动态
新一代测序技术的出现为生命科学重大问题研究提供
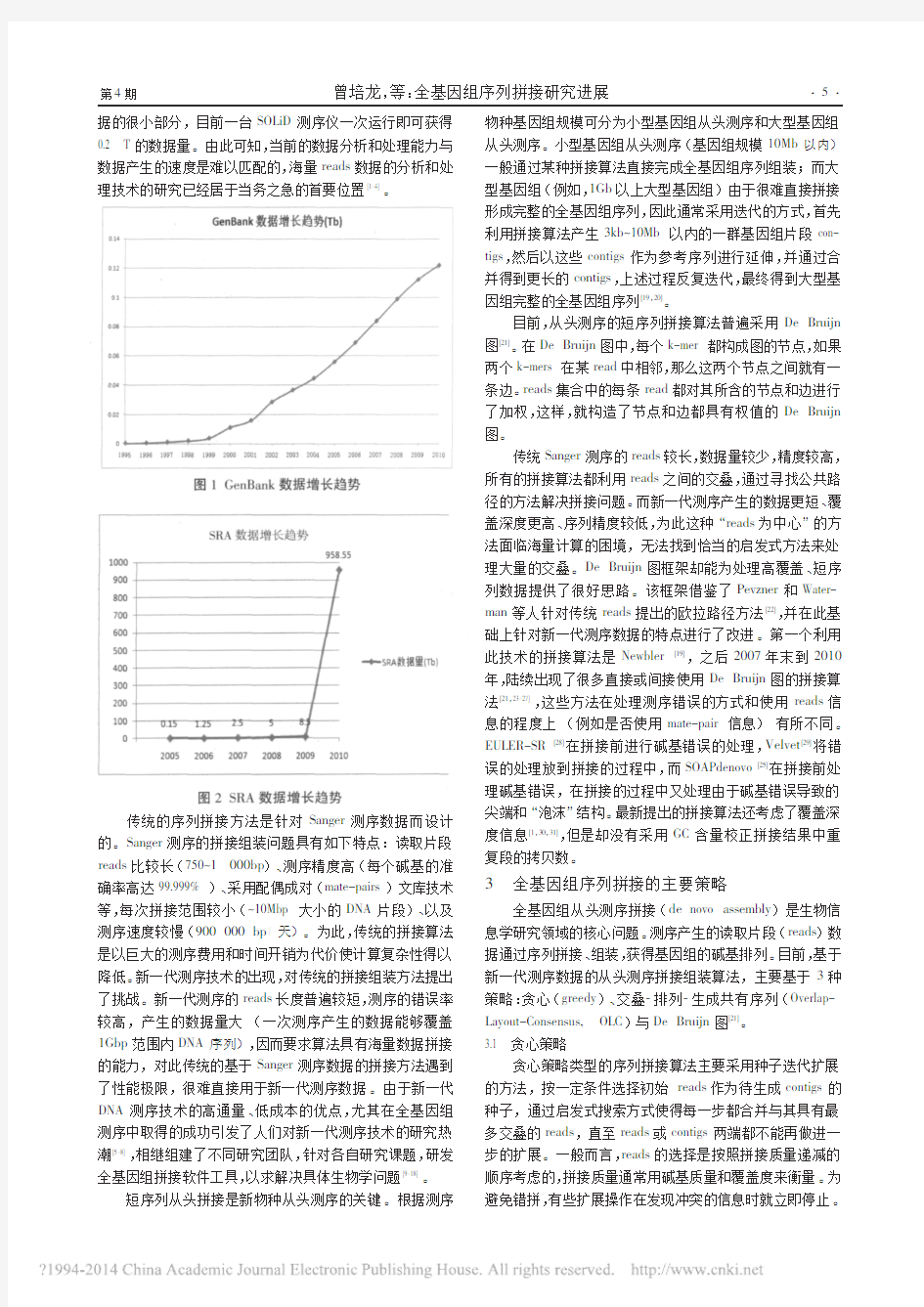
新的手段的同时,其海量数据及其长度短、精度相对较低等特点,为生物信息学设置了前所未有的时代挑战。海量reads 数据的处理能力远远落后于测序数据的爆炸性增长速度,测试数据的快速、准确分析已经成为生命科学研究的短板[3]。如图1所示,从2006~2010年积累的新一代短片段数据量远远超过了过去10年所获得的基因组测序数据的总和。
符合SRA 标准的新一代测序数据从2005~2010年的增长情况如图2所示。与图1相比可以看出,数据分析速度远远落后于数据产生速度,尤其是2010年数据的增长更是属于
“爆炸式的”,而这些还只占目前产生的新一代测序数全基因组序列拼接研究进展
(哈尔滨工业大学计算机科学与技术学院,哈尔滨150001)
摘要:全基因组序列拼接是生物信息学研究领域的核心问题。针对新一代测序数据读取片段reads 长度短、
数据海量、精确度低等特点带来的严峻挑战,能够满足实际应用的序列拼接软件的研发,已成为生物信息学领域最为迫切的研究课题。深入探讨全基因组序列拼接的发展动态、所采用的主要策略等方面,总结序列拼接相关理论,并为未来新算法的研发提出具体的改进建议。
关键词:中图分类号:TP391
文献标识码:A
文章编号:2095-2163(2012)04-0004-05
Research Progress of Whole Genome Assembly
ZENG Peilong,WANG Yadong
Abstract :Whole genome assembly is the core issue of bioinformatics.On conditions that next generation sequencing brings bioinfor-
matics an unprecedented challenge due to its data of mass,short length and relatively low precision,development of sequence assembly soft-ware that could meet practical application has become the most important research topic.This paper analyses the development progress and main strategies of whole genome assembly deeply,sums up the relevant theory and provide specific suggestions for future algorithms.
Key words:全基因组序列拼接;生物信息学;新一代测序
Whole Genome Assembly ;Bioinformatics ;Next-Generation Sequencing
(School of Computer Science and Technology,Harbin Institute of Technology,Harbin 150001,China )
曾培龙,王亚东
智能计算机与应用
INTELLIGENT COMPUTER AND APPLICATIONS
Vol.2No.4第2卷第4期2012年8月
Aug.2012
据的很小部分,目前一台SOLiD测序仪一次运行即可获得0.2T的数据量。由此可知,当前的数据分析和处理能力与数据产生的速度是难以匹配的,海量reads数据的分析和处理技术的研究已经居于当务之急的首要位置[1-4]。
传统的序列拼接方法是针对Sanger测序数据而设计的。Sanger测序的拼接组装问题具有如下特点:读取片段reads比较长(750~1000bp)、测序精度高(每个碱基的准确率高达99.999%)、采用配偶成对(mate-pairs)文库技术等,每次拼接范围较小(~10Mbp大小的DNA片段)、以及测序速度较慢(900000bp/天)。为此,传统的拼接算法是以巨大的测序费用和时间开销为代价使计算复杂性得以降低。新一代测序技术的出现,对传统的拼接组装方法提出了挑战。新一代测序的reads长度普遍较短,测序的错误率较高,产生的数据量大(一次测序产生的数据能够覆盖1Gbp范围内DNA序列),因而要求算法具有海量数据拼接的能力,对此传统的基于Sanger测序数据的拼接方法遇到了性能极限,很难直接用于新一代测序数据。由于新一代DNA测序技术的高通量、低成本的优点,尤其在全基因组测序中取得的成功引发了人们对新一代测序技术的研究热潮[5-8],相继组建了不同研究团队,针对各自研究课题,研发全基因组拼接软件工具,以求解决具体生物学问题[9-18]。
短序列从头拼接是新物种从头测序的关键。根据测序物种基因组规模可分为小型基因组从头测序和大型基因组从头测序。小型基因组从头测序(基因组规模10Mb以内)一般通过某种拼接算法直接完成全基因组序列组装;而大型基因组(例如,1Gb以上大型基因组)由于很难直接拼接形成完整的全基因组序列,因此通常采用迭代的方式,首先利用拼接算法产生3kb~10Mb以内的一群基因组片段con-tigs,然后以这些contigs作为参考序列进行延伸,并通过合并得到更长的contigs,上述过程反复迭代,最终得到大型基因组完整的全基因组序列[19,20]。
目前,从头测序的短序列拼接算法普遍采用De Bruijn 图[21]。在De Bruijn图中,每个k-mer都构成图的节点,如果两个k-mers在某read中相邻,那么这两个节点之间就有一条边。reads集合中的每条read都对其所含的节点和边进行了加权,这样,就构造了节点和边都具有权值的De Bruijn 图。
传统Sanger测序的reads较长,数据量较少,精度较高,所有的拼接算法都利用reads之间的交叠,通过寻找公共路径的方法解决拼接问题。而新一代测序产生的数据更短、覆盖深度更高、序列精度较低,为此这种“reads为中心”的方法面临海量计算的困境,无法找到恰当的启发式方法来处理大量的交叠。De Bruijn图框架却能为处理高覆盖、短序列数据提供了很好思路。该框架借鉴了Pevzner和Water-man等人针对传统reads提出的欧拉路径方法[22],并在此基础上针对新一代测序数据的特点进行了改进。第一个利用此技术的拼接算法是Newbler[19],之后2007年末到2010年,陆续出现了很多直接或间接使用De Bruijn图的拼接算法[21,23-27],这些方法在处理测序错误的方式和使用reads信息的程度上(例如是否使用mate-pair信息)有所不同。EULER-SR[28]在拼接前进行碱基错误的处理,Velvet[29]将错误的处理放到拼接的过程中,而SOAPdenovo[25]在拼接前处理碱基错误,在拼接的过程中又处理由于碱基错误导致的尖端和“泡沫”结构。最新提出的拼接算法还考虑了覆盖深度信息[1,30,31],但是却没有采用GC含量校正拼接结果中重复段的拷贝数。
3全基因组序列拼接的主要策略
全基因组从头测序拼接(de novo assembly)是生物信息学研究领域的核心问题。测序产生的读取片段(reads)数据通过序列拼接、组装,获得基因组的碱基排列。目前,基于新一代测序数据的从头测序拼接组装算法,主要基于3种策略:贪心(greedy)、交叠-排列-生成共有序列(Overlap-Layout-Consensus,OLC)与De Bruijn图[21]。
3.1贪心策略
贪心策略类型的序列拼接算法主要采用种子迭代扩展的方法,按一定条件选择初始reads作为待生成contigs的种子,通过启发式搜索方式使得每一步都合并与其具有最多交叠的reads,直至reads或contigs两端都不能再做进一步的扩展。一般而言,reads的选择是按照拼接质量递减的顺序考虑的,拼接质量通常用碱基质量和覆盖度来衡量。为避免错拼,有些扩展操作在发现冲突的信息时就立即停止。
曾培龙,等:全基因组序列拼接研究进展·5·第4
期
SSAKE[16]、SHARCGS[11]、VCAKE[9]即采用了该类拼接策略。SSAKE和VCAKE能够处理非完全匹配的reads,SHARCGS 适用于均匀分布、非配对的reads。贪心策略适用于小型基因组,而对于有大量重复序列存在的大型基因组的测序数据进行拼接时,拼接效果往往很差。
3.2交叠-排列-生成共有序列(OLC)策略
OLC策略在第一代测序中被广泛采用,并取得了很好的结果。该种策略主要包含3个主要的步骤:
(1)构建交叠图,计算任意两条reads之间的交叠。为了减少计算复杂度,可以先对reads建立类似后缀数据、后缀树的索引,而后在所建索引的基础上进行计算;
(2)排列reads,确定reads之间的相对位置,建立ove-rlap图,分析overlap图,获得遍历整个图的最佳近似路径;
(3)生成共有序列,通过多序列比对等方法,获得最终的基因组序列。
由于新一代测序数据的reads海量,计算reads交叠的平方复杂度以及reads长度较短等限制,基于OLC策略的拼接方法并不适于处理新一代的海量短序列数据,为此,在该种策略的基础上又相继提出了多个更加实用的拼接算法,主要有:CABOG[32]、Edena[12]、Shorty[33]。Shorty用于处理SOLiD数据,利用300-500bp长度的种子上的配对数据,采用类似于Huson等人[34]的方法,来估算两个相邻contigs之间的gap 的大小。CABOG基于Celera Assembler[14],采用一种被称为“rocks and stones”的技术,先通过reads之间的交叠关系,建立reads之间的多序列比对,然后使用配对数据分割不满足约束条件的多序列比对,再由多序列比对上的配对数据确定其相对位置,最终生成共有序列。
随着测序技术的不断发展,基因组测序产生的数据质量会越来越高,生成的reads片段也会越来越长,以reads为计算中心的拼接策略或许会再次进入人们的视野,成为研究主题。
3.3De Bruijn图策略
基于De Bruijn图(DBG)策略(如图3所示)的拼接算法被最广泛地应用到新一代测序数据的处理中。典型算法有:ABySS[10]、ALLPATHS[23]、Euler-SR[28]、SOAPdenovo[25]和Velvet[29]。基于De Bruijn图的拼接算法,非常巧妙地将具有交叠关系的reads映射到一起,降低了计算交叠时的复杂度,减少了内存消耗。基于De Bruijn图策略的拼接算法的大致步骤是:
(1)构建De Bruijn图。将reads分割成一系列连续的子串k-mers(一般用K值表征kmer碱基数目的大小),作为图中的边,相邻的两个k-mers交叠(K-1)个碱基;
(2)化简De Bruijn图。方法是合并路径出度入度唯一的节点,按照一定的规则去除图中的尖端(tips)和泡状结构(bubbles);
(3)构建contigs。在De Bruijn图或其子图中寻找一条最优的欧拉路径(一次且仅有一次地经过每条边的路径),该路径对应的碱基序列即为contigs;
(4)生成scaffolding。利用配对数据,确定contigs之间的相对方向与位置关系,对contigs进行组装,并填充con-tigs之间的gaps,最终得到scaffolds序列。
基于De Bruijn图的拼接算法中,一个关键操作是K值的选择。选择大的K值能够解决更多的短小重复片段(tiny repeats),降低图的复杂性,但同时也降低了图的连通性,后续的拼接过程会产生更多的间隙(gaps);选择小的K值,对应的De Bruijn图具有相对好的连通性,但图变得更加复杂,重复片段的处理也变得更加困难,增加了错拼的可能性。目前,还没有通用的K值选择方法,需要根据特定的应用,选择合适的K值。一般认为对于原核生物的基因组拼接,K值选取在21~35之间是合适的[25,29];而对于真核生物基因组的K值的选择要相对复杂得多,目前还没有明确的结论或者一致的建议。
3.4序列拼接算法的比较
自从基因组测序产生以来,序列拼接算法就不断地处于研发和改进之中。Hernande等人[12]的研究表明,通常,基于图的拼接算法与采用贪心策略的拼接算法相比,在序列长度和准确率,运行时间以及内存消耗等方面,往往具有相对更好的拼接表现。基于OLC策略的拼接算法多用于传统测序数据的拼接,而基于De Bruijn图的拼接算法则更多地用于新一代测序数据。不同的拼接算法在处理不同的测序数据时,通常具有各异的表现,目前还没有一种拼接程序能在所有方面都表现得出色,这个结论可在Narzisi等人[35]的研究成果中找到论据:由于基因组和测序数据的复杂性,拼接长度与准确率往往是一个平衡的关系,高精度往往是以牺牲长度为代价的,反之亦然。而这种平衡如何选择,则取决于具体的应用。同样,拼接结果的准确率与算法的内存消耗也存在类似的平衡关系。就适用的基因组规模而言,除了SOAPdenovo[25]、AByss[10]等少数软件外,大多数拼接软件只适用于简单的小型基因组。目前,几乎所有软件都需要较大内存的计算平台。如何优化数据处理方法、高效地存储海量reads数据,是序列拼接算法软件研发过程中必须面对的一个重要课题。
4全基因组序列拼接的发展方向
目前,绝大多数的基因组拼接软件还无法满足具体的应用需要,无论在拼接质量、拼接效率还是内存消耗等方面都需要更进一步地改进和完善。展望未来,全基因组的序列拼接组装存在着如下几个发展方向:
(1)采用多种配对文库(mate-pair或paired-end)。基因组中存在的大量重复片段是序列拼接过程中最难解决的
第2卷
智能计算机与应用·6
·
问题,也是影响拼接质量的关键因素。配对数据可以为重复片段的解决提供有利助益:大片段配对文库用于解决大尺寸的重复片段,小片段配对文库用于解决小尺寸的重复片段。结合不同片段大小的配对文库,能够起到相互补充的作用;
(2)使用长序列(Sanger数据或contigs)与短序列的混合数据策略。长序列片段具有很好的片段交叠性,使拼接的可信度增强,并能跨越小的重复片段;而短序列具有很高的覆盖度,能够减少并填充拼接过程中出现的序列间隙(gaps)。长短序列的结合使用,能有效避免错误拼接,提升拼接质量;
(3)并行化拼接。基因组的序列拼接并行化包括两个方面:一是reads数据的分布式存储;二是拼接过程的并行化。reads数据的分布式存储能有效解决基因组拼接所需大型计算平台的问题,有助于内存瓶颈的缓解。拼接过程的并行化能加快序列的拼接速度,提高拼接效率。因此,并行化是基因组序列拼接软件发展的一个主要趋势。
5结束语
新一代测试数据在提高测序速度、降低测序成本的同时,也给后续的生物信息学数据处理带来了严峻的挑战。尽管目前人们针对各自的研究课题开发了一些处理全基因组序列拼装软件,并且成功地进行了许多物种的全基因组拼接。然而,数据的处理和分析过程一般局限在大型集群的计算平台上完成,并且拼接质量较低、效率也不高(小型基因组也需要若干小时的拼接组装时间),迫切需要适合于新一代测序的海量reads数据拼接算法,以便为新一代测序数据的分析与处理提供理论指导和技术支持,加快全基因组测序研究的推进。
参考文献:
[1]METZKER M L.Sequencing Technologies-the Next Generati-
on[J].Nat Rev Genet,2010,11(1):31-46.
[2]SHENDURE J,JI H.Next-generation DNA sequencing[J].Nat-
Biotechnol,2008,26(10):1135-1145.
[3]POP M,SALZBERG S L.Bioinformatics challenges of new se-
quencing Technology[J].Trends Genet,2008,24(3):142-149.[4]WHITEFORD N,HASLAM N,WEBER G,et al.An analysis of
the feasibility of short read sequencing[J].Nucleic Acids Res,2-005,33(19):e171.
[5]WANG J,WANG W,LI R,et al.The diploid genome sequence
of an Asian individual[J].Nature,2008,456(7218):60-65.[6]LI R Q,LI Y R,ZHENG H C,et al.Building the sequence
map of the Human pan-genome[J].Nature Biotechnology,2010,
28(1):57-63.
[7]TONG P,PRENDERGAST J G,LOHAN A J,et al.Sequencing
and analysis of an Irish human genome[J].Genome Biol,2010,
11(9):R91.
[8]HIATT J B,PATWARDHAN R P,TURNER E H,et al.Paral-
lel,tag-directed assembly of locally derived short sequence rea-ds[J].Nat Methods,2010,7(2):119-122.
[9]JECK W R,REINHARDT J A,BALTRUS D A,et al.Extend-
ing assembly of short DNA sequences to handle error[J].Bioinf-ormatics,2007,23(21):2942-2944.
[10]SIMPSON J T,WONG K,JACKMAN S D,et al.Abyss:a p-
arallel assembler for short read sequence data[J].Genome Res, 2009,19(6):1117-1123.
[11]DOHM J C,LOTTAZ C,BORODINA T,et al.Sharcgs,a fast
and highly accurate short-read assembly algorithm for De Novo genomic sequencing[J].Genome Research,2007,17(11):16-97-1706.
[12]HERNANDEZ D,FRANCOIS P,FARINELLI L,et al.De No-
vo bacterial genome sequencing:millions of very short reads assembled on a desktop computer[J].Genome Research,2008, 18(5):802-809.
[13]DIGUISTINI S,LIAO N Y,PLATT D,et al.De Novo genom-
e sequence assembly o
f a filamentous fungus Usin
g Sanger,454
and Illumina Sequence Data[J].Genome Biology,2009,10(9): R94.
[14]LI R Q,FAN W,TIAN G,et al.The sequence and De Novo
assembly of the Giant panda genome[J].Nature,2010,463(7-279):311-317.
[15]YE K,SCHULZ M H,LONG Q,et al.Pindel:a pattern gro-
wth approach to detect break points of large deletions and m-edium sized insertions from paired-end short reads[J].Bioinfor-matics,2009,25(21):2865-2871.
[16]WARREN R L,SUTTON G G,JONES S J,et al.Assembling
millions of short DNA sequences using ssake[J].Bioinformatics, 2007,23(4):500-501.
[17]ZIMIN A V,SMITH D R,SUTTON G,et al.Assembly reco-
nciliation[J].Bioinformatics,2008,24(1):42-45.
[18]HOSSAIN M S,AZIMI N,SKIENA S.Crystallizing short-read
assemblies around seeds[J].BMC Bioinformatics,2009,10(Su-ppl+1):S16.
[19]FLICEK P,BIRNEY E.Sense from sequence Reads:methods
for alignment and assembly.Nat Methods,2009,6(11Suppl): S6-S12.
[20]LI R Q,LI Y R,KRISTIANSEN K,et al.Soap:short oligon-
ucleotide alignment program[J].Bioinformatics,2008,24(5):7-13-714.
[21]MILLER J R,KOREN S,SUTTON G.Assembly algorithms for
next-generation sequencing data[J].Genomics,2010,95(6): 315-327.
[22]PEVZNER P A,TANG H,WATERMAN M S.An Eulerian p-
ath approach to DNA fragment assembly[C]//Proc Natl Acad Sci USA,2001,98(17):9748-9753.
[23]BUTLER J,MACCALLUM I,KLEBER M,et al.Allpaths:De
Novo assembly of whole-genome shotgun Microreads[J].Genome Research,2008,18(5):810-820.
[24]MACCALLUM I,PRZYBYLSKI D,GNERRE S,et al.All pa-
ths2:small genomes assembled accurately and with high con-tinuity from short paired Reads[J].Genome Biology2009,10(10):R103.
曾培龙,等:全基因组序列拼接研究进展·7·第4期
(下转第13页)
[7]王显志,王忠杰,徐晓飞.面向服务整合的顾客个性化需求
分类与描述方法[J].黑龙江大学自然科学学报,2009,26(5):622-626.[8]
莫同,徐晓飞,王忠杰.面向服务系统设计的服务需求模型[J].
计算机集成制造系统,2009,15(4):661-669.
[9]詹蓉,陈荣秋.个性化需求分类的定量分析研究[J].软科学,
2007,21(3):5-8.
[25]LI R,ZHU H,RUAN J,et al.De Novo assembly of human
genomes with massively parallel short Read sequencing[J].Ge-nome Research,2009,20(2):265-272.[26]CHAISSON M J,BRINZA D,PEVZNER P A.
De Novo frag-
ment assembly with short mate-paired Reads:Does the Read length matter芽[J].Genome Research,2009,19(2):336-346.[27]SCHEIBYE-ALSING K,HOFFMANN S,FRANKEL A,et al.
Sequence assembly[J].Comput Biol Chem,2009,33(2):121-136.
[28]CHAISSON M J,PEVZNER P A.Short Read fragment assem-
bly of Bacterial genomes[J].Genome Research,2008,18(2):324-330.
[29]ZERBINO D R,BIRNEY E.Velvet:algorithms for De Novo
short read assembly using De Bruijn graphs[J].Genome Resea-rch,2008,18(5):821-829.
[30]REINHARDT J A,BALTRUS D A,NISHIMURA M T,et al.
De Novo assembly using low-coverage short Read sequence d-ata from the rice pathogen pseudomonas Syringae Pv.Oryzae.[J]Genome Research,2009,19(2):294-305.
[31]HARISMENDY O,NG P C,STRAUSBERG R L,et al.Eval-
uation of next generation sequencing platforms for population targeted sequencing studies[J].Genome Biology,2009,10(3):R32.
[32]MILLER J R,DELCHER A L,KOREN S,et al.Aggressive
assembly of pyrosequencing reads with mates[J].Bioinformatics,2008,24(24):2818-2824.
[33]HOSSAIN M S,AZIMI N,SKIENA S.Crystallizing short-read
assemblies around seeds[J].BMC Bioinformatics,2009,10(Su-(ppl+1):S16.
[34]HUSON D H,REINERT K,MYERS E W.The greedy path-
merging algorithm for contig scaffolding[J].Journal of the Acm,2002,49
(5):603-615.[35]MILLER J R,DELCHER A L,KOREN S,et al.Aggressive
assembly of pyrosequencing Reads with mates[J].Bioinformati-cs,2008,24(24):2818-2824.
[36]HOSSAIN M S,AZIMI N,SKIENA S.Crystallizing short-read
assemblies around seeds[J].BMC Bioinformatics,2009,10(Su-ppl+1):S16.
[37]NARZISI G,MISHRA B.Comparing De Novo genome assemb-
ly:the long and short of It[J].PLoS ONE,2011,6(4).
闫昕,等:支持大规模个性化需求描述的服务过程模型
第4期
·13·
(上接第7页)
..
....................................................................................................................................
42 SeqMan 笔记本:A电脑 创建时间:2013/12/10 8:35更新时间:2013/12/10 9:07 1.打开lasergene-dnastart-seqman 2.点击add sequences,注意文件格式为.ab1,该文件为测序峰图文件。 3.添加序列文件,本例为16_xxxx.ab1,点击打开,序列添加到Selected sequences窗口。 4.点击done,序列成功加入主程序窗口 5.选中想要拼接的序列,点击assemble,拼接开始。 6.拼接完成后出现,拼接成功提示,creating new contig1:from xxx entering xxx
7.点击窗口右上角,“-”最小化,将拼接提示最小化,回到主窗口。 8. 此时主窗口上方出现拼接好的contig1的信息,574bp,来源于两条序列。 9.双击contig1出现具体的拼接过程窗口。 10.点击16前的黑色三角符号,可以看到序列峰图(注意峰图非常重要,不同颜色代表不同碱基,峰型表示测序可信度)。 11.详细讲一下峰图: 测序反应开始时和结束时的序列是读不准的(测序的原理决定)。一个测序反应最多能测定500-800个碱基,且测序反应开始和结束的碱基读不准。
ITS45的长度在500bp左右,意味着单向测序末端会读不准。 采用双向测序,在R向峰分辨率极度降低时,F向 正好处在分辨率最高的测序区域,所以这段序列程序会以F向测序结果为准。 seqman在序列拼接的同时,让测序峰图可见,让我们可以判断测序结果的可靠性。 12.接着说拼接完成后如何拷贝拼接好的序列,其实非常简单,选中顶上的consensus中的序列,全选,ctrl+C,拼接好的序列就复制到剪切板中了,可以粘贴到txt中使用。
发表于《中国奶牛》,2011 全基因组选择育种技术及在奶牛育种中应用进展 范翌鹏1孙东晓1* 张勤1张胜利1张沅1刘林2 (1.中国农业大学动物科技学院,北京,100193; 2.北京奶牛中心. 北京. 100085) 摘要:全基因组选择是指基于基因组育种值(GEBV)的选择方法,指通过检测覆盖全基因组的分子标记,利用基因组水平的遗传信息对个体进行遗传评估,以期获得更高的育种值估计准确度。由于可显著缩短世代间隔,全基因组选择作为一种育种新技术在奶牛育种中具有广阔的应用前景,目前已经成为各国的研究热点。不同国家的试验结果表明,在奶牛育种工作,基于GEBV 的遗传评估可靠性在20-67%之间,如果代替常规后裔测定体系,可节省92%的育种成本。本文综述了全基因组选择的基本原理及其在各国奶牛育种中的应用现状和所面临的问题。 关键词:全基因组选择,奶牛育种 Genome-Wide Selection and its Application in Dairy Cattle FAN YiPeng, SUN Dongxiao, ZHANG Qin, ZHANG Shangli, ZHANG Yuan, LIU Lin (College of Animal Science Technology, China Agricultural University, Beijing, 100193) Abstract: Genomic selection refers to selection decisions based on genomic breeding values (GEBV). The GEBV are calculated as the sum of the effects of dense genetic markers, or haplotypes of these markers, across the entire genome, thereby potentially capturing all the quantitative trait loci (QTL) that contribute to variation in a trait. Genomic selection has become a focus of study in many countries as the new breeding method. Reliabilities of GEBV for young bulls without progeny test results in the reference population were between 20 and 67%. By avoiding progeny testing, bull breeding companies could save up to 92% of their costs [1]. In this paper, we first review the progress of genomic selection, including the principle, methods, accuracy and advantages of genomic selection. We then review the application of genomic selection in dairy cattle. Key words: Genomic Selection, Dairy Breeding 全基因组选择(Genomic Selection,GS),即全基因组范围的标记辅助选择(Marker Assisted Selection, MAS),指通过检测覆盖全基因组的分子标记,利用基因组水平的遗传信息对个体进行遗传评估,以期获得更高的育种值估计准确度。研究已表明,标记辅助选择可提高奶牛育种遗传进展[2][3],但是在目前奶牛育种工作中却无法大规模推广应用标记辅助选择。因为奶牛的生产性状和健康性状均受大量基因座位共同影响,通过有限数量的已知标记无法大幅度加快遗传进展;其次,通过精细定位策略鉴定主效基因需花费大量人力物力和时间;而且利用标记信息估计育种值的计算方法也很复杂。全基因组选择基于基因组育种值(Genomic Estimated Breeding Value, GEBV)进行选择,其实施包括两个步骤:首先在参考群体中使用基因型数据和表型数据估计每个染色体片段的效应;然后在候选群体中使用个体基因型数据估计基因组育种值(genomic breeding value,GEBV)[4],模拟研究证明,仅仅通过标记预测育种值的准确性可以达到0.85(指真实育种值与估计育种值之间的相关,而可靠性则指其平方)。如果在犊牛刚出生时即可达到如此高的准确性,对奶牛育种工作则具有深远意义。模拟研究表明:对于一头刚出生的公犊牛而言,如果其GEBV的估计准确性可以达到经过后
全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中 数以百万计的单核苷酸多态性(single nucleotide ploymorphism ,SNP)为分子 遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。 随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。 全基因组关联方法首先在人类医学领域的研究中得到了极大的重视和应用,尤其是其在复杂疾病研究领域中的应用,使许多重要的复杂疾病的研究取得了突破性进展,因而,全基因组关联分析研究方法的设计原理得到重视。 人类的疾病分为单基因疾病和复杂性疾病。单基因疾病是指由于单个基因的突变导致的疾病,通过家系连锁分析的定位克隆方法,人们已发现了囊性纤维化、亨廷顿病等大量单基因疾病的致病基因,这些单基因的突变改变了相应的编码蛋白氨基酸序列或者产量,从而产生了符合孟德尔遗传方式的疾病表型。复杂性疾病是指由于遗传和环境因素的共同作用引起的疾病。目前已经鉴定出的与人类复杂性疾病相关联的SNP位点有439 个。全基因组关联分析技术的重大革新及其应用,极大地推动了基因组医学的发展。(2005年, Science 杂志首次报道了年龄相关性视网膜黄斑变性GWAS结果,在医学界和遗传学界引起了极大的轰动, 此后一系列GWAS陆续展开。2006 年, 波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的GWAS结果(Herbert 等. 2006);2007 年, Saxena 等多个研究组联合报道了与2 型糖尿病( T2D ) 关联的多个位点, Samani 等则发表了冠心病GWAS结果( Samani 等. 2007); 2008 年, Barrett 等通过GWAS发现了30 个与克罗恩病( Crohns ' disrease) 相关的易感位点; 2009 年, W e is s 等通过GWAS发现了与具有高度遗传性的神经发育疾病——自闭症关联的染色体区域。我国学者则通过对12 000 多名汉族系统性红斑狼疮患者以及健康对照者的GWAS发现了5 个红斑狼疮易感基因, 并确定了4 个新的易感位点( Han 等. 2009) 。截至2009 年10 月, 已经陆续报道了关于人类身高、体重、 血压等主要性状, 以及视网膜黄斑、乳腺癌、前列腺癌、白血病、冠心病、肥胖症、糖尿病、精神分 裂症、风湿性关节炎等几十种威胁人类健康的常见疾病的GWAS结果, 累计发表了近万篇 论文, 确定了一系列疾病发病的致病基因、相关基因、易感区域和SNP变异。) 标记基因的选择: 1)Hap Map是展示人类常见遗传变异的一个图谱, 第1 阶段完成后提供了 4 个人类种族[ Yoruban ,Northern and Western European , and Asian ( Chinese and Japanese) ] 共269 个个体基因组, 超过100 万个SNP( 约1
序列拼接 * 为了保证测序结果的准确性,单基因短片段(700pd左右)测序一般应采用双向测序,然后将双向测序的结果拼接在一起,从而获得一致性序列。线粒体基因组测序和DNA长片段测序一般是通过分段测序来完成的,最后也需要将测出的短片段拼接成一条完整的序列。序列拼接可以在不同的软件中进行。 一、使用“组装批处理文件byLHM.pg4”进行拼接 1. 在预定的位置建立一个文件夹“gap”,将需要使用的3个软件“组装批处理文件byLHM.pg4”、“V ector_primer4pMD18-T.vec_pri”、“pMD18-T_Vector.seq”拷贝到该文件夹下,再将需要拼接的测序文件拷贝到该文件夹下。 2. 双击运行“组装批处理文件byLHM.pg4”程序。 3. 在程序运行后出现的界面右侧点击“Add files”按钮,打开要拼接的序列文件。为了保证 拼接后输出的是正向序列,最好先添加上游引物序列,然后添加下游引物序列,因为在一般情况下软件将添加的第一条序列默认为正向参照序列;有时由于测序效果等因素的影响,有时即使首先添加的是上游引物序列,但拼接后仍然会以测序效果明显更好的下游引物序列为正向参照序列,此时需要按照后面介绍的方法将上游引物序列转换为正向参照序列再输出一致性序列。 4. 点击界面上方第二行的“Configure Modules”,在弹出的窗口左边的任务栏中点击“[x] Sequencing vector Clip”,再点击右边的“Browse”按钮,通过弹出的窗口打开“Vector_primer4pMD18-T.vec_pri”程序;点击左边任务栏中的“[] Cloning Vector Clip”,再点击右边的“Browse”按钮,通过弹出的窗口打开“pMD18-T_Vector.seq”程序;点击左下角的“Run”按钮,即开始数据处理,处理结果将自动保存到“gap”文件夹中。 5. 在“gap”文件夹中双击“AssMit_tmp.o.aux”文件,将鼠标移到弹出的“Contig Selector” 窗口中的直线上,点击右键,选择“Edit Contig”,即弹出“Contig Editor”窗口,点击最右边的“setting”按钮,在下拉菜单中选择“By background colour”,即可显示比对结果的有差异碱基;双击某一序列,即可显示该序列的测序峰图,以检查核对该位点碱基的测序情况。 * 注:执行此操作时一定要检查正向序列是否为上游引物序列;如果不是,则需要将上游引物序列转换成正向序列后再执行下面的“输出及保存序列”操作;具体的操作步骤是:点击“GAPv4.10 AssMit_tmp.o”窗口中的“Edit”菜单,在下拉菜单中选择“Complement a contig”命令,在弹出来的“Complement contig”小窗口中检查确认“Contig identifier” 框中的序列为上游引物序列,然后点击“OK”即将完成序列转换。 6. 点击“GAPv4.10 AssMit_tmp.o”窗口中的“File”菜单,在下拉菜单中选择“Save consensus”可保存一致序列,nomors------ok ,序列即保存在刚刚使用过的那个文件夹中,然后把文件名改成用“*.txt”形式,以便保存的文件成为文本文件,若忘记在文件名后加“.txt”,则保存完毕后可将文件的扩展名改成“.txt”;只有拼接好的一致序列才可用于后面的序列分析。 7.然后把在ncbi里查到的相近种的序列放到一起,也可以直接放到刚才那个cons.txt文本文 档中,然后打开clustalx.exe进行序列比对,file------load sequence ------G盘-----004文件夹-----cons.txt-----aligenment-----do complete aligenment,这时如果发现两条序列的保守区域很不对,极可能是刚刚测得这个种的序列反了,需要用Bioedit把它正过来, 8.在程序里打开已经安装好的Bioedit,例如找file---------open----G盘---004----cons.txt,打开, 选sequence--------下拉菜单中找Nuclic acid,在菜单中找reverse complement,点击它 然后在另一对话框中例如G:/004/CONS.TXT中点击保存save Aligenment. 这样序列即
中国水产科学 2011年7月, 18(4: 936?943 Journal of Fishery Sciences of China 综述 收稿日期: 2011?03?14; 修订日期: 2011?04?10. 基金项目: 国家自然基金资助项目(30730071; 30972245; 农业科技成果转化资金项目(2010GB24910700. 作者简介: 于洋(1987?, 硕士研究生. E-mail: yuy8866@https://www.doczj.com/doc/cb14321440.html, 通信作者: 张晓军, 副研究员. E-mail: xjzhang@https://www.doczj.com/doc/cb14321440.html, DOI: 10.3724/SP.J.1118.2011.00935 全基因组选择育种策略及在水产动物育种中的应用前景 于洋1,2 , 张晓军1 , 李富花1 , 相建海1 1. 中国科学院海洋研究所实验海洋生物学重点实验室, 山东青岛266071; 2. 中国科学院研究生院, 北京 100049 摘要: 全基因组选择的概念自2001年由Meuwissen 等提出后便引起了动物育种工作者的广泛关注。目前, 澳大利亚、新西兰、荷兰、美国的研究小组已经应用该方法进行了优质种牛的选择育种, 并取得了很好的效果。此外在鸡和猪的选择育种中也有该方法的应用, 但在水产动物选育中尚未见该方法使用的报道。本文对“全基因组选择育种”的概念和提出背景进行了归纳, 对全基因组选择育种的优势进行了阐述, 并详细介绍了其具体的策略, 总结了目前全基因组育种所广泛采用的方法以及取得的成果, 旨在为该方法在水产动物育种方面的应用研究提供科学参考。 关键词: 全基因组选择; 水产动物育种; SNP; QTL; 全基因组育种值估计 中图分类号: S96 文献标志码: A 文章编号: 1005?8737?(201104?0935?08 人类对于动物的选择育种由来已久, 最初所进行的只是简单的人工驯化。随着遗传学研究的发展, 尤其是“数量遗传学理论”的提出, 动物育种技术进入快速发展时
如何查找基因序列?(转载) (2010-08-01 11:47:41) 如何查找基因序列? ——在Genbank中寻找目的基因的实例 ——献给受类似问题困扰的广大酷友,以及给我动力和信心发表原创帖的基因酷的朋友们。 酷友感言:网络的世界很精彩,网络的查询很无奈。为了我们的科学研究事业,为了我们能够顺利毕业,我们的广大酷友们在网络的海洋里遨游…遨游…咋就找不到彼岸呢?今天要设计这个基因的PCR引物,明天又要查那个基因的信息,那么大一张网,唉想起来就郁闷……鉴此,我们推出了利用Genbank查找基因序列的帖子,希望对大家有所帮助,并请大家多多指教!当然,如果您已经是此中高手,那就权当我是班门弄斧了,呵呵。 1. 根据文献 搞reasearch肯定要读文献的,如果你曾经在文献中看到过你感兴趣的基因,而且文中还提到了该基因在Genbank中的ID号,那就好办了,直接打开https://www.doczj.com/doc/cb14321440.html,,在Search后的下拉框中选择Nucleotide,把Genbank ID号输入GO前面的文本框中,点“GO”,就可以找到他了。 举例说明,例如:在2003年JBC的文章(Conditional Knock-out of
Integrin-linked Kinase Demonstrates an Essential Role in Protein Kinase B/Akt Activation)中出现了“calreticulin (GenBank accession number gi 16151096)”,那么把“16151096”输入GO前面的文本框中,点“GO”,就可以找到该基因了(当然包括基因序列等相关信息)。 在出现了检索结果界面(下图)后,直接点击红箭头所指的 AY047586就可以看到基因的相关信息了...(呵呵,是不是有点太......easy 了) 这里需要指出一下,在显示基因的页面右侧有一个Link,点击后出现一个小菜单,里面是与该基因相关的链接,很有用的,值得一个一个地去看看,这里我就不多说了。点击 AY047586后出现的界面如下:如果你只想获得序列(例如去设计PCR引物的时候),那就可以选择FASTA,这样就得到了FASTA格式的序列文件,没有其他数字和格式的干扰。 (缩略图,点击图片链接看原图)这就是FASTA格式的序列: (缩略图,点击图片链接看原图)2. 根据已经获得的基因的相关信息进行查找(待续......) 鼓励一下吧,累坏了正如路漫漫所说,如果只是知道基因的名字,怎么查序列呢?还是举例说明,比如我想做的基因名称是人的VEGF基因,那么怎么在Genbank中找到它呢?还是一步一步来...打开https://www.doczj.com/doc/cb14321440.html,/ 在search后面的下拉框中选择Gene,然后在中间的文本框中输入基
王前飞: (1)为什么要研究表观遗传学? 答: 表观遗传学主要通过DNA 的甲基化、组蛋白修饰、染色质重塑和非编码RNA 调控等方式控制基因表达。表观遗传学是近几年兴起的而且发展迅速的一个研究遗传的分支学科,其研究和应用不仅对基因表达、调控、遗传有重要作用,而且在肿瘤、免疫等许多疾病的发生和防治以及干细胞定向分化研究、基因芯片中亦具有十分重要的意义。表观遗传学补充了“中心法则”忽略的两个问题,即哪些因素决定了基因的正常转录和翻译以及核酸并不是存储遗传信息的唯一载体;在分子水平上,表观遗传学解释了DNA序列所不能解释的诸多奇怪的现象。如: 同一等位基因可因亲源性别不同而产生不同的基因印记疾病,疾病严重程度也可因亲源性别而异。表观遗传学信息还可直接与药物、饮食、生活习惯和环境因素等联系起来,营养状态能够通过改变表观遗传以导致癌症发生,尤其是维生素和必需氨基酸。 此外,表观遗传学信息的改变,对包括人体在内的哺乳动物基因组有广泛而重要的效应,如转录抑制、基因组印记、细胞凋亡、染色体灭活等。DNA 甲基化模式的改变,尤其是某些抑癌基因局部甲基化水平的异常增加,在肿瘤的发生和发展过程中起到了不容忽视的作用。研究发现,肿瘤细胞DNA 存在广泛的低甲基化和局部区域的高甲基化共存现象,以及总的甲基化能力增高,这3个特征各以不同的机制共同参与甲基化在肿瘤发生、发展中的作用。如胃癌、结肠癌、乳腺癌、肺癌、胰腺癌等众多恶性肿瘤都不同程度地存在一个或多个肿瘤抑制基因CpG 岛甲基化。而表观遗传学改变在本质上的可逆性,又为肿瘤的防治提供了新的策略。所以,随着表观遗传学研究的深入,肯定会对人类生长发育、肿瘤发生以及遗传病的发病机制及其防治做出新的贡献,也必将在其他领域中展示其不可估量的作用和广阔的前景。 (2)表观遗传学涉及到哪些方面? 答: 表观遗传学的研究内容主要包括:DNA甲基化、组蛋白的末端修饰和变异体、DNAaseⅠ高敏感位点、非编码RNA、转录因子及其辅助因子、顺式调控元件和基因组印记等。 (3)什么因素会影响基因表达水平? 答: 基因选择性转录表达的调控( DNA甲基化,基因印记,组蛋白共价修饰,染色质重塑) 基因转录后的调控(基因组中非编码RNA,微小RNA(miRNA),反义RNA、内含子、核糖开关等) 1.转录水平的调控:包括DNA转录成RNA时的是否转录及转录频率的调控,DNA 的序列决定了DNA的空间构型,DNA的空间构型决定了转录因子是否可以顺利的结合到DNA的调控序列上,比如结合到TATA等序列上。 2.翻译水平的调控:翻译水平的调控又可以分成翻译前的调控和翻译后的调控。 a、翻译前的调控主要是RNA编辑修饰。 b、翻译后调控主要是蛋白的修饰,蛋白修饰后可以成为有功能的蛋白或者有隐藏功能的蛋白。 在真核和原核细胞中,从基因表达到蛋白质合成,其间有许多地方受到调控,这
2014年成都理工大学校内数学建模竞赛论文 二0一四年五月二十五日
摘要:本文所要研究的就是全基因组的从头测序的组装问题。 首先,本文简要介绍了测序技术及测序策略,认真分析了基因系列拼装所面临的主要挑战,比如reads数据海量、可能出现的个别碱基对识别错误、基因组中存在重复片段等复杂情况,探讨了当前基因组序列拼接所采用的主要策略,即OLC(Overlap/Layout/Consensus)方法、de Bruijn图方法,且深入探讨了de Bruijn图方法。 其次,针对题中问题,以一条reads为基本单位,分为reads拼接和contig组装两个阶段,其中contig是由reads拼接生成的长序列片段。Reads的拼接阶段主要包括数据预处理、de-Bruijn 图、contig构建等,而contig的组装阶段主要包括序列的相对位置的确定以及重叠部分overlap的检测,用序列比对的方法来提高拼接的精度。 最后,进行了算法的验证与性能的评价,并且针对问题2,进行了组装分析与验证,结果表明,得到的拼接基因组序列在小范围内与原基因组序列大致吻合。 关键词:基因组系列拼接; reads;de Bruijn图;contig组装;k-mer片段;
一.问题重述 基因组组装 快速和准确地获取生物体的遗传信息对于生命科学研究具有重要的意义。对每个生物体来说,基因组包含了整个生物体的遗传信息,这些信息通常由组成基因组的DNA或RNA分子中碱基对的排列顺序所决定。获得目标生物基因组的序列信息,进而比较全面地揭示基因组的复杂性和多样性,成为生命科学领域的重要研究内容。 确定基因组碱基对序列的过程称为测序(sequencing)。测序技术始于20世纪70年代,伴随着人类基因组计划的实施而突飞猛进。从第一代到现在普遍应用的第二代,以及近年来正在兴起的第三代,测序技术正向着高通量、低成本的方向发展。尽管如此,目前能直接读取的碱基对序列长度远小于基因组序列长度,因此需要利用一定的方法将测序得到的短片段序列组装成更长的序列。通常的做法是,将基因组复制若干份,无规律地分断成短片段后进行测序,然后寻找测得的不同短片段序列之间的重合部分,并利用这些信息进行组装。例如,若有两个短片段序列分别为 ATACCTT GCTAGCGT GCTAGCGT AGGTCTGA 则有可能基因组序列中包含有ATACCTT GCTAGCGT AGGTCTGA这一段。当然,由于技术的限制和实际情况的复杂性,最终组装得到的序列与真实基因组序列之间仍可能存在差异,甚至只能得到若干条无法进一步连接起来的序列。对组装效果的评价主要依据组装序列的连续性、完整性和准确性。连续性要求组装得到的(多条)序列长度尽可能长;完整性要求组装序列的总长度占基因组序列长度的比例尽可能大;准确性要求组装序列与真实序列尽可能符合。 利用现有的测序技术,可按一定的测序策略获得长度约为50–100个碱基对的序列,称为读长(reads)。基因组复制份数约为50–100。基因组组装软件可根据得到的所有读长组装成基因组,这些软件的核心是某个组装算法。常用的组装算法主要基于OLC(Overlap/Layout/Consensus)方法、贪婪图方法、de Bruijn 图方法等。一个好的算法应具备组装效果好、时间短、内存小等特点。新一代测序技术在高通量、低成本的同时也带来了错误率略有增加、读长较短等缺点,现有算法的性能还有较大的改善空间。 问题一:试建立数学模型,设计算法并编制程序,将读长序列组装成基因组。你的算法和程序应能较好地解决测序中可能出现的个别碱基对识别错误、基因组中存在重复片段等复杂情况。 问题二:现有一个全长约为120,000个碱基对的细菌人工染色体(BAC),采用Hiseq2000测序仪进行测序,测序策略以及数据格式的简要说明见附录一和附录二,测得的读长数据见附录三,测序深度(sequencing depth)约为70×,即基因组每个位置平均被测到约70次。试利用你的算法和程序进行组装,并使之具有良好的组装效果。 附录一:测序策略 测序策略如下图所示。DNA分子由两条单链组成,在图中表现为两条平行直
全基因组选择在猪育种上的研究进展 自野生动物被驯化以来,科学家一直致力于提高畜禽育种值的研究。近半个世纪来,畜禽育种值估计的方法主要经历了综合选择指数法、同期群体比较法、最佳线性无偏预测法(Best LinearUnbiased Prediction,BLUP)、分子标记辅助选择育种(MAS)以及近几年快速发展的GS 法。同时,随着高密度基因芯片的出现和高通量测序技术的快速发展,单核苷酸多态性(SingleNucleotide Polymorphism,SNP)分型成本快速下降,GS 才逐渐引起畜禽界的关注。特别是Schaeffer发现,在奶牛育种中利用GS比后裔测定可节约成本97%,且遗传进展可提高3~4倍后,全球掀起了一股研究GS的热潮。 全基因组选择(GS) 什么是GS 2001年,Meuwissen等人最先提出GS,实质为全基因组范围的标记辅助选择。其理论基础是应用整个基因组的标记信息和各性状值来估计每个标记或染色体片段的效应值,然后将效应值加和即得到基因组育种值(GenomicEstimated Breeding Value,GEBV)。GS在某种程度上是MAS的延伸,弥补了在MAS 中标记数量只能解释一部分遗传方差以及数量性状位点(QuantitativeTrait Locus,QTL) 定位困难的缺点。其中心任务是提高GEBV值的准确性,并尽可能准确地估计每个标记的效应。而估计标记效应的方法在实际运用中以BLUP法为主;Bayes法虽其准确性高于BLUP,但因其计算复杂,需在超级计算机上运行而限制其应用。不过随着快速算法的开发和计算机硬件的改进,Bayes法的运算效率有望提高。 为什么选用GS GS的优势 与MAS相比,GS的优势主要表现在: 1)能对所有的遗传和变异效应做出准确的估计。而MAS 只能对部分遗传变异进行检测,且容易高估其遗传效应。 2)缩短世代间隔、提高畜禽年遗传进展、降低生产成本等,这在需要后裔测定的家畜中尤为明显。如GS给奶牛育种带来了巨大经济效益。 3)早期选择准确率高。 4)对于较难实施选择的性状具有重大影响。如低遗传力性状、难以测定的性状等。 5)GS在提高种群的遗传进展前提下,还能降低群体的近交增量。 GS的可靠性
全基因组从头测序(de novo测序) https://www.doczj.com/doc/cb14321440.html,/view/351686f19e3143323968936a.html 从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动物、植物、细菌、真菌的全基因组序列,从而推进该物种的研究。一个物种基因组序列图谱的完成,意味着这个物种学科和产业的新开端!这也将带动这个物种下游一系列研究的开展。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。华大科技利用新一代高通量测序技术,可以高效、低成本地完成所有物种的基因组序列图谱。包括研究内容、案例、技术流程、技术参数等,摘自深圳华大科技网站 https://www.doczj.com/doc/cb14321440.html,/service-solutions/ngs/genomics/de-novo-sequencing/ 技术优势: 高通量测序:效率高,成本低;高深度测序:准确率高;全球领先的基因组组装软件:采用华大基因研究院自主研发的SOAPdenovo软件;经验丰富:华大科技已经成功完成上百个物种的全基因组从头测序。 研究内容: 基因组组装■K-mer分析以及基因组大小估计;■基因组杂合模拟(出现杂合时使用); ■初步组装;■GC-Depth分布分析;■测序深 度分析。基因组注释■Repeat注释; ■基因预测;■基因功能注释;■ ncRNA 注释。动植物进化分析■基因家族鉴定(动物TreeFam;植物OrthoMCL);■物种系统发育树构建; ■物种分歧时间估算(需要标定时间信息);■基因组共线性分析; ■全基因组复制分析(动物WGAC;植物WGD)。微生物高级分析 ■基因组圈图;■共线性分析;■基因家族分析; ■CRISPR预测;■基因岛预测(毒力岛); ■前噬菌体预测;■分泌蛋白预测。 熊猫基因组图谱Nature. 2010.463:311-317. 案例描述 大熊猫有21对染色体,基因组大小2.4 Gb,重复序列含量36%,基因2万多个。熊猫基因组图谱是世界上第一个完全采用新一代测序技术完成的基因组图谱,样品取自北京奥运会吉祥物大熊猫“晶晶”。部分研究成果测序分析结果表明,大熊猫不喜欢吃肉主要是因为T1R1基因失活,无法感觉到肉的鲜味。大熊猫基因组仍然具备很高的杂合率,从而推断具有较高的遗传多态性,不会濒于灭绝。研究人员全面掌握了大熊猫的基因资源,对其在分子水平上的保护具有重要意义。 黄瓜基因组图谱黄三文, 李瑞强, 王俊等. Nature Genetics. 2009. 案例描述国际黄瓜基因组计划是由中国农业科学院蔬菜花卉研究所于2007年初发起并组织,并由深圳华大基因研究院承担基因组测序和组装等技术工作。部分研究成果黄瓜基因组是世界上第一个蔬菜作物的基因组图谱。该项目首次将传
此名为ContigExpress的软件可用于做序列拼接,主要使用方法如下: 1.解压缩下载的压缩文件contig.zip文件,保证文件CExpress.exe,Gexudat.def在同一个目录下,打开Cexpress.exe应用程序,进入ContigExpress操作界面,如图1。 图1 2.点击菜单上的“Project”选择“Add Fragments”,一般我们发给您的是AB1文件,如果您有其它格式的文件,也可以选择,在这里我们选择AB1文件,以其为例,如图2。 图2 3.选择您存放AB1文件(即我们Email给您的测序结果的彩图文件)的目录,选择文件类型为ALL FILES, 之后打开要拼接的AB1,从而添加进ContigExpress软件。在此以A、B 两个序列为例,如果有多个序列的也可以同时添加进入。
图3 4.选中要拼接的序列,再选菜单“Assemble”栏下的“Assemble Selected Fragments”命令,或用工具栏上的按钮,如图3。若两个结果能够拼接起来的,会得到一个Assemble1下的contig1的结果,如图4。 图4 5.双击contig1,打开拼接后的结果,选中菜单“VIEW”栏,进入VIEW OPTION,将SHOW ALIGNMENT AS 由TEXT 改为GRAPH.,点击OK 后得到结果如图5。此时可能会因为两条序列的测序结果误差,会有不同的地方,在拼接图片框中的绿色竖杠就表示了这些不同的地方,如图所示。接着可点击绿色竖杠找到有误差的地方,进行修改。 6.在修改过程中,遇到有误差的地方,可以根据峰形来判断是多读还是漏读来进行修改,此时电脑认为是漏读碱基的地方会以点来表示,如图5,此处很明显是A序列上多读了一个G碱基,可将其删除。(注:因为软件本身的问题,只有在拼接过程中是正向的序列才能进行修改操作,若在反向上修改碱基,保存时会产生错误而直接关闭程序。所以若要修改反向序列上的碱基,可先保存后,把原有的Assemble1的结果拆开,点序列图标上的“Name”,如图3,所选中的序列上的一个“name”横栏,使序列按Name的升降次序来排列,把要作为正向的序列放到要作为反向序列上面即可。以此序列为例,将其改变方向后可实现反
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。 3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:
利用SeqMan进行序列拼接 Step1:打开Seqman软件 Step2:加入你要拼接的序列 点击Add sequences 查找并选中要拼接的序列(可按住control键进行多选) 点击Add按钮填加选择的序列 填加完后点击done 注:最好用测序的图谱尽量不要直接用测序得到的序列 Step3:去除末端序列 主要是去除序列末端测序质量差或是载体序列 有两种方法可以用来去除这类末端序列 其一:利用Seqman自带的去除工具自动去除(利用Trim ends按钮进行) 其二:手工去除 个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示 手工去除侧翼序列 双击要去除侧翼序列的目标序列 将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接
此步请将测序不准确或认为是载体的序列用这种方法去除。 测序准确的峰形图 峰形规则,一般在序列的中部,如下图所示 测序不准确的峰形图 峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示
Step4:进行序列拼接 点击Assemble按钮 在新出现窗口处点击拼接好的contig1 在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图 Step5:查找拼接错误 find conflict 点击菜单Edit 点击Find Previous或Find Next查找接接中出现的错误 还可以通过Seqman左下角的快捷按钮查找错误的拼接
关于内参基因的选择 实验内参,即是在检测细胞内分子表达变化时选择的参照物,其在细胞内的表达相对恒定,在处理因素作用条件下不会发生表达改变的基因。内参同样可以校正上样量、上样过程中存在的实验误差,保证实验结果的准确性。 1、管家基因 最普通的内参是内源性参照基因,也就是管家基因(持家基因,house keeping gene)。 管家基因是一类始终保持着低水平的甲基化并且一直处于活性转录状态的基因,高度保守并且在大多数情况下持续表达。其表达水平受环境因素影响较小,而且是在个体各个生长阶段的大多数,或几乎全部组织中持续表达,或变化很小,因此常存在于生物细胞核的常染色质中。它的表达只受启动序列或启动子与RNA 聚合酶相互作用的影响,而不受其他机制调节。 管家基因维持细胞最低限度功能所不可少的基因, 如编码组蛋白基因、编码核糖体蛋白基因、线粒体蛋白基因、糖酵解酶的基因等。这类基因在所有类型的细胞中都进行表达,因为这些基因的产物对于维持细胞的基本结构和代谢功能是必不可少的。
2、内参基因选择的条件 1、不存在假基因,以免基因组DNA的扩增; 2、高度或中度表达,避免太高或太低的丰度; 3、稳定表达于不同类型的细胞和组织中,表达量无明显差异; 4、表达水平与细胞周期、活化等无关; 5、不受外源性或内源性因素的影响。 3、不同管家基因 在选择管家基因作为内参时,首先要按不同类型的分子选择正确的内参。曾看到有人用检测miRNA时选择了GAPDH作为内参呢。 a、检测mRNA时的内参 通常使用的是GAPDH、beta-actin、tubulin GAPHD
GAPDH GAPDH是糖酵解反应中的一个酶,由4个30-40kDa的亚基组成,分子量 146kDa。该酶基因为管家(house keeping)基因,几乎在所有组织中都高水平表达,在同种细胞或者组织中的蛋白质表达量一般是恒定的,且不受含有的部分识别位点、佛波脂等的诱导物质的影响而保持恒定,故被广泛用作抽提total RNA,poly(A)+ RNA,Western blot等实验操作的标准化的内参。 beta-actin β-Actin是PCR常用的内参,β-Actin抗体是Western Blot很好的内参指数。β-Actin是横纹肌肌纤维中的一种主要蛋白质成分,也是肌肉细丝及细胞骨架微丝 的主要成分,具有收缩功能,分布广泛。
基因组序列的差异分析 ----mVISTA的在线使用说明 当然,除了在线版的,我们还可以在网站上填写信息申请离线的软件。但我试用了一下,需要先自己比对,然后要按照一定的格式来制作文件,当然你还必须得安装java才能运行软件;总之,我感觉没有在线版的方便。 1 将数据放入服务器中 在首页,你将被要求确定你想要分析的基因组序列的数量。输入这个数字之后,点击“提交”,将带你到主提交页面。 mVISTA服务器最多可以同时处理100条序列。 1.1主提交页面必填的内容 E-mail 地址 通过E-mail,我们可以提示你的在线处理已经得到结果。
序列 你可以用2种方式来上传你的序列: 1.使用“Browse”按钮从你的电脑上,上传纯文本的Fasta格式文件。如果是一个作为参 考的生物体的DNA序列必须作为一个contig提交(可以进行一定的定向排列将多个片段合并为一个contig),而其他非参考序列可以在一个或多个contig中提交(draft)。 Fasta格式的示例序列(您可以在NCBI站点上找到关于该格式的更多细节): >mouse ATCACGCTCTTTGTACACTCCGCCATCTCTCTCT … !!!注意:序列里面我们只接受字母CAGTN和X。请确保提交序列是作为一种纯文本格式,而不是Word或HTML文件格式。 如果您以FASTA格式提交序列,我们建议您为它取一个有意义的名称(比如直接是你的物种名之类的),因为这些名称将出现在我们生成的图形中。如果您使用的是一个draft草图序列,那么结果中每个contigs的命名都将按照您在“>”符号后指示的命名进行。 2.您可以给出它的GenBank登录号,系统将自动从GenBank数据库里进行检索序列。 在这两种情况下,序列的总大小都不应超过10M,而且任何一条序列都不应超过2M。 1.2主提交页面选填的内容 这些选项允许您自定义您的VISTA分析。您可以使用独立获得的基因注释,选择合适的Repeat Masker选项,给分析的序列指定名称,并改变序列保存分析的参数。如果您没有填写这些选填选项,我们将使用它们的默认值。 比对程序 根据您分析的具体内容(参见“about”-链接中的详细信息),您可以选择以下比对程序之一:1、AVID----全局两两比对。如果您选择使用这个程序,其中一个序列应该被完成比对,其他 所有序列可以完成或以草图draft格式完成。对于集合中所有已完成的序列,AVID生成所有相对所有成对的比对结果,可以使用任何序列作为基础(参考)来显示。如果某些序列是草图格式,AVID将生成它们与最终序列的比对,这将被用作基础(参考)。这是该服务器上唯一可以处理草图序列的比对程序。 (小知识:草图序列与完整序列DNA sequence, draft: Sequence of a DNA with less accuracy than a finished sequence. In a draft sequence, some segments are missing or are in the wrong order or are oriented incorrectly. A draft sequence is as opposed to a finished DNA sequence.)2、LAGAN----完成完整序列的全局两两比对和多重比对。如果某些序列是草图格式,您的查 询将被重定向到AVID以获得两两比对。多重比对将由VISTA可视化,它将计算并显示序列的保守区,以您指示的任何序列作为参考。这是该服务器上唯一能够产生真正的多重