
1 引言
1.1 苯并咪唑的性质简介
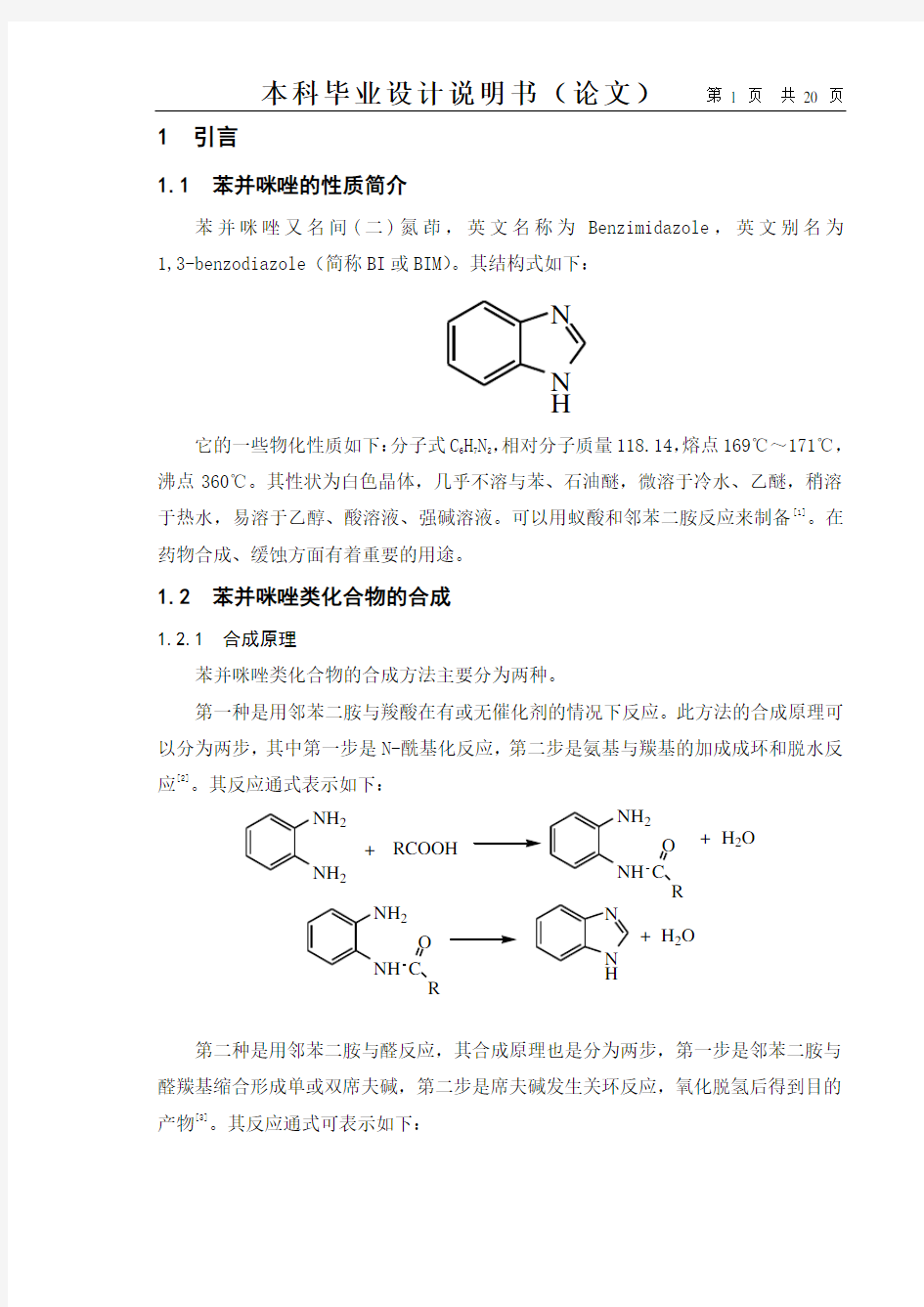
苯并咪唑又名间(二)氮茚,英文名称为Benzimidazole,英文别名为1,3-benzodiazole(简称BI或BIM)。其结构式如下:
H
N
它的一些物化性质如下:分子式C
6H
7
N
2
,相对分子质量118.14,熔点169℃~171℃,
沸点360℃。其性状为白色晶体,几乎不溶与苯、石油醚,微溶于冷水、乙醚,稍溶于热水,易溶于乙醇、酸溶液、强碱溶液。可以用蚁酸和邻苯二胺反应来制备[1]。在药物合成、缓蚀方面有着重要的用途。
1.2 苯并咪唑类化合物的合成
1.2.1 合成原理
苯并咪唑类化合物的合成方法主要分为两种。
第一种是用邻苯二胺与羧酸在有或无催化剂的情况下反应。此方法的合成原理可以分为两步,其中第一步是N-酰基化反应,第二步是氨基与羰基的加成成环和脱水反应[2]。其反应通式表示如下:
NH2
NH2
RCOOH
NH
NH2
C
R
O
+
+H2O NH
NH2
C
R
O
H
N
+H2O
第二种是用邻苯二胺与醛反应,其合成原理也是分为两步,第一步是邻苯二胺与醛羰基缩合形成单或双席夫碱,第二步是席夫碱发生关环反应,氧化脱氢后得到目的产物[3]。其反应通式可表示如下:
R-CHO N
NH 2H N
H
R NH 2
NH
2
+N H
N
R
1.2.2 苯并咪唑类化合物的合成方法
如上所述,用羧酸和邻苯二胺反应来合成苯并咪唑类化合物是两种方法之一。如,付红蕾等[4]以邻苯二胺和丙酸为原料,磷酸为催化剂,合成出了2-乙基苯并咪唑。其原料的摩尔比是1:2.5,反应时间为2.0h ,反应温度约为100℃,磷酸用量为1.5mL 时,最终得率为78.09%。
另外,王元有[5]研究了在多聚磷酸和五氧化二磷催化下,邻苯二胺分别与对苯二甲酸、间苯二甲酸反应合成出了两种苯并咪唑衍生物,产率均在80%以上。并且经过了红外光谱,紫外可见光谱的初步表征,还定性研究了它们的荧光性质。其合成路线如下:
RCOOH PPA/P O NH 2
NH 2
+NH NH 2O
PPA/P O H
N
R
与此同时,利用醛类和邻苯二胺的反应来合成苯并咪唑类化合物也已多见于文献。如李焱等[6]用醛和邻苯二胺为原料,在碘催化下,一锅法合成出了2-取代苯并咪唑,该方法具有催化剂毒性低,反应条件温和,反应时间较短,且产率较高(73%~78%)等特点。
于丽颖等[7]研究了邻苯二胺和苯甲醛反应,在空气氧化下,得出了合成2-苯基苯并咪唑的简便方法。并且得出了在原料的摩尔比为1:2,反应的时间为3h ,反应温度在44℃时,氧化产物的产率可以达到72.50%,她们宣称这个方法后处理比较简单,对设备腐蚀性也较小,是一条符合现代环保要求的优良工艺路线。
李莹莹等[8]
用邻苯二胺与芳香醛反应,并且在不加任何催化剂的情况下用空气作
为氧化剂,甲醇为溶剂,合成出了苯并咪唑的衍生物,且产率均超过69%。其反应方程式如下:
NH2
NH2
ArCH2O
H
N
H
Ar
air O
N
NH2
C Ar +
2
其中Ar=4-CH
3C6H
4
, -3-ClC
6
H
4
,-BrC
6
H
4
,2-NO
2
C
6
H
4
,4-Cl
2
C
6
H
3
,5-Cl-2-NO
2
C
6
H
5
,
4-Cl
2C
6
H
5
。
除此之外,也有其他方法见诸文献。如陈兴权等[9]在NaOH催化下,用邻苯二胺和氨基氰合成出了2-氨基苯并咪唑,并得出了较佳的工艺条件。
王济奎等[10]用甲醛与邻硝基苯胺反应,用TiO
2
为催化剂和自主研制的微型光催化反应器一步法合成出了苯并咪唑,且产率较高。合成路线如下:
+HCHO TiO2
H
N
N
NH2
NH2
另外,他们还以Ti为催化剂,相同条件下合成出了苯并咪唑,并得出了最佳工艺条件[10,11]。
刘思全等[12]还以A2甘氨酸和邻苯二胺作为原料,合成出了2-氨基烷基苯并咪唑的衍生物:2-甲胺基苯并咪唑。
1.3 苯并咪唑类化合物的应用
1.3.1苯并咪唑类化合物在药物方面的应用
据文献报道,苯并咪唑类化合物作为杀菌剂的主要代表就是多菌灵[13]和苯菌灵[14],并且这两种药物在经过了40多年的发展后仍占有相当的市场份额。其机理为植物病原与β-微蛋白相结合,从而破坏其功能,以此来抑制病原菌的有丝分裂和形态建构。由于其应用时间长已经产生抗性,研究人员在在多菌灵的基础上开发出了多菌灵磷酯[15],其杀菌范围和杀菌效果与多菌灵相差不大,但合成成本有明显的降低[16]。
在抗寄生虫方面的应用。噻苯达唑是第一代抗寄生虫药物,因为其毒性太大已被淘汰[16]。现在,苯并咪唑氨基甲酸酯是发展相当迅速且药效良好的广谱抗虫药物,其现在已经成为大部分寄生蠕虫疾病治疗的最佳选择[17,18]。
除此之外,在质子泵抑制剂方面、在抗动脉粥样硬化活性、抗病毒、组胺抗拮剂
等方面,苯并咪唑类化合物也有重要的用途[18]。
1.3.2 苯并咪唑类化合物在缓蚀方面的应用
众所周知在化工等工业部门中,各种反应器在清洗时经常采用的是酸洗的方法,但是因为金属材料在酸洗的时候会出现不同的程度的腐蚀,所以我们经常要加入适量的缓蚀剂来降低金属在酸性条件下的腐蚀速率,从而提高酸洗效果。苯并咪唑类缓蚀剂以其较低的毒性(大鼠经口半致死量>10000mg/Kg)[19]而得到广泛使用,属于环境友好型缓蚀剂,有较好的市场前景,因此大量的研究者在开发新的产品并探索工艺优化条件。
文献报道,苯并三氮唑是铜的特效缓蚀剂,但因其毒性较大,对环境危害严重,已逐渐被其他缓蚀剂所替代。并且已经有大量的研究证明,苯并咪唑类化合物是可以作为黄铜的缓蚀剂的,且效果极佳。另外,他们还对其缓蚀机理作了研究说明。
胡莲跃等[20]研究了黄铜在3%NaCl溶液中,温度为30℃时,苯并咪唑对其的缓蚀行为。结果表明,在苯并咪唑的质量浓度为0.4g/L的情况下其缓蚀效果可高达93.3%。并且还得出结论:随着温度的升高苯并咪唑的缓蚀能力是呈下降趋势的。另外他们还对75℃下苯并咪唑在黄铜表面的吸附行为进行了研究,研究表明苯并咪唑在黄铜表面的吸附是服从郎缪尔吸附等温式的自发过程的,属于物理吸附。
史志龙等[21]测定了两种烷基苯并咪唑:2-己基苯并咪唑和2-十一基苯并咪唑在浓度为0.5mol/L的HCl溶液中对黄铜的缓蚀速率。并且得到它们的缓蚀率分别为48.4%、54%。其中2-十一基苯并咪唑的缓蚀率要大于2-己基苯并咪唑。并且还得出烷基苯并咪唑在铜表面的吸附为物理吸附,是符合Freudlich吸附等温式。
王清华等[22]对苯并咪唑衍生物添加剂的抗腐蚀性能进行了考察,同时还对腐蚀后铜表面的有机膜进行了分析,并对它们的抗腐蚀机理进行了探究。结果表明:苯并咪唑衍生物添加剂的抗腐蚀机理为成膜型。
在碳钢缓蚀方面的作用。研究者[23]对苯并咪唑(BI),2-甲基苯并咪唑(2-CH
3
-BI),
2-巯基苯并咪唑(2-SH-BI),2-氨基苯并咪唑(2-NH
2
-BI),四种缓蚀剂在浓度为1mol/L 的HCl溶液中对A3钢的缓蚀性能进行了研究。结果表明,四种缓蚀剂均具有较好的缓
蚀性能,且缓蚀率从大到小依次为2-SH-BI>2-NH
2-BI>2-CH
3
-BI>BI。四种缓蚀剂在A3
钢表面发生了物理、化学混合吸附,其规律服从郎缪尔吸附等温式。另外,沈建等[24]探究了20#碳钢在含有苯并咪唑类化合物的HCl溶液中的腐蚀行为。结果发现在室温和50℃的条件下,5种苯并咪唑化合物(苯并咪唑、2-丙基苯并咪唑、2-对氯苄基苯并
咪唑、2-戊基苯并咪唑、2-己基苯并咪唑)的缓蚀效果都是非常明显的[24]。其中2-对氯苄基苯并咪唑的缓蚀性能是它们中最好的(缓蚀率可达97%)。
在锌材缓蚀方面的应用。胡莲跃等[25]研究了苯并咪唑在浓度为0.1moL/L的KOH溶液中对锌材的缓蚀性能。结果表明:苯并咪唑可以有效地抑制锌的阳极氧化,从而抑制锌在碱液中的自腐蚀,属于阳极型缓蚀剂,并且当苯并咪唑的浓度为10.0mmoL/L时缓蚀的效果是最佳的,其缓蚀率可高达96.68%,因此我们可以说该方法可以取代汞作为锌电极的缓蚀剂,并且有利于环境保护,为工业部门又提供了一种行之有效的缓蚀途径。
1.4 本论文研究内容及其方法
综上所述,苯并咪唑及其衍生物是具有较高的药用价值和缓蚀性能的,并且市场前景非常好。尤其是在缓蚀方面它们具有对环境友好,毒性低,合成工艺简单,对设备要求不高,合成工艺成熟等特点。所以本论文研究了苯并咪唑的合成及其应用。
综合所查阅的文献及实验室现有条件,本次研究采用邻苯二胺与甲酸反应,并在磷酸的催化下来合成苯并咪唑。用单一因素法去探索该反应的最佳工艺条件,即邻苯二胺和甲酸的摩尔配比、催化剂种类、催化剂的用量、反应的温度、反应的时间,以及溶液pH对反应得率的影响。同时,对合成出来的产物利用其自身的物理化学性质,显微熔点仪和红外光谱进行初步的检测,来观察是否与文献所述的相关值与特征相符合,以验证所合成出来的产物是苯并咪唑。
在苯并咪唑的应用方面。主要是根据GB10124-88金属材料实验室均匀腐蚀全浸试验方法 [26]来研究苯并咪唑的缓蚀作用。其方法为将标准A3(Q235)钢板(尺寸为50mm×25mm×2mm),经打磨、冲洗、脱脂,干燥后分别浸入含有不同浓度苯并咪唑的6moL/L的HCl溶液中,并且在室温下持续浸没72h后,通过失重法来衡量各浓度下苯并咪唑的腐蚀效率,从而来评价苯并咪唑的缓蚀性能。
2 实验部分
2.1 实验所需试剂与材料
表2.1 实验中所需的试剂
名称分子式纯度生产厂家
邻苯二胺C6H8N2CP(化学纯) 国药集团化学试剂有限公司甲酸HCOOH AR(分析纯) 无锡市佳妮化工有限公司磷酸H3PO4AR(分析纯) 上海中试化工总公司
盐酸HCl AR(分析纯) 无锡市佳妮化工有限公司无水乙醇CH3CH2OH AR(分析纯) 国药集团化学试剂有限公司磷钨酸H3O40PW12·X H2O AR(分析纯) 国药集团化学试剂有限公司
另外本实验在测定缓蚀性能时用的是A3(Q235)钢板,其尺寸为50mm×25mm×2mm。2.2 实验所需的仪器
表2.2 实验中所需的仪器
名称型号生产厂家数显恒温油浴锅HH-1S 金坛市杰瑞尔电器有限公司恒温水浴锅HH-1 国华电器有限公司
分析天平AL104 梅特勒-托利多仪器(上海)有
限公司
电子天平HC-2102 慈溪市华徐衡器实业有限公司新型电热恒温鼓风干燥箱DHG-9240 上海康路仪器设备有限公司精密显微熔点测定仪X-5 北京福凯仪器有限公司
精密增力电动搅拌器JJ-1 国华电器有限公司
红外分光光度计TJ270-30A 天津市拓普仪器有限公司
玻璃仪器有:四口烧瓶(250mL)、烧杯(1000mL,50mL)、容量瓶(250mL)、量筒(5mL,50mL)、温度计(0℃~200℃)、玻璃棒、玻璃漏斗、试剂瓶、抽滤瓶,
表面皿。
其他还有:铁架台、布氏漏斗、压片机、玛瑙坩埚、红外烘箱、热吹风机、冰柜、试
管夹、乳胶吸管、洗瓶、药匙、滤纸、pH试纸、称量纸、砂纸,剪刀。2.3 实验的合成步骤
本实验利用邻苯二胺与甲酸反应,以磷酸或磷钨酸作催化剂来合成苯并咪唑。其典型的合成步骤如下:
(1)准备阶段。将四口烧瓶与精密增力电动搅拌器固定在一起,并将四口烧瓶放入数显恒温油浴锅中,调节浸入程度与搅拌桨位置,确保可轻松转动并能起到较好的搅拌作用。
(2)根据所设计条件,用电子天平称取相应重量的邻苯二胺与甲酸,用量筒量取相应体积的催化剂后,放入四口烧瓶中,然后打开数显恒温油浴锅的温度调节开关,调节至所需温度,同时打开精密增力电动搅拌器开始搅拌,等到温度升至所需温度时开始计算搅拌反应时间。此时需注意要用温度计测量油浴锅的显示温度是否与实际温度相符合。
(3)调节溶液的pH。待反应完毕,将烧瓶抬起,等到烧瓶内溶液的温度自然冷却至室温后,再将溶液倒入50mL烧杯中,并用事先配制好的10%的氢氧化钠溶液调节反应所得溶液的pH至9~10,此时溶液中会有白色固体析出。
(4)简单过滤。将玻璃漏斗固定在铁架台上,放入菊花形滤纸,润湿并调整。然后将上步所得溶液缓慢倒入玻璃漏斗内,待滤干后再用少量的蒸馏水去洗涤滤饼。注意在溶液不黄时可省略此步骤,因为苯并咪唑是微溶于冷水的。
(5)重结晶。将此上得到的滤饼移入250mL烧杯中,并用洗瓶冲洗滤纸上的残留滤饼,随后加入适量的水,并用表面皿盖住烧杯口,防止沸水过量蒸发。然后放入已加热到100℃的恒温水浴锅中进行重结晶操作。在此之间为了进行给所得固体脱色,需要放入适量的活性炭,但需注意的是在加活性炭时要给体系降温,以防止溶液暴沸冲料,损失产品[27]。
(6)趁热过滤。待固体全部溶解后,为了防止温度迅速下将有晶体析出要趁热过滤。此时,可将布氏漏斗放入沸水中蒸煮一段时间,然后迅速放入烧杯中,并将烧杯放入恒温水浴锅内,以保证过滤时的温度。此时迅速将溶液倒入布氏漏斗内进行常压过滤,并覆盖表面皿保持漏斗内温度。过滤后再在水浴锅中加热一段时间,如此便可以得到较好的晶体形态。第一次过滤如有残留的晶体在滤纸上要重新进行加热过滤,直至无残留为止。并将过滤后的溶液先放在室温下缓慢冷却,使晶体逐渐生长,然后再进行冰水浴,使结晶较完全。
(7)抽滤。将冷却析出晶体的溶液进行抽滤,抽滤时为了减少损失,应用滤液来洗涤残留在烧杯壁上的晶体,不可用水来冲洗。抽滤结束时要先将滤瓶与水泵间相连的皮管拔去,再关闭水泵,以防止水倒流入滤瓶内。
(8)完成以上步骤后,将得到的晶体压紧后放入烘箱内干燥,并控制烘箱到适宜的温度。待烘干后计算产物的得率,并保留以用作熔点实验和红外光谱测试的样品。
2.4 产物的表征方法
2.4.1 熔点法
本实验采用X-5型精密显微熔点测定仪来测定苯并咪唑的熔点范围。用显微熔点仪表征的优点是它可以检测微量样品和高熔点样品,并且可以精确的观察到样品受热熔化时的全部过程。检测步骤如下:
(1)准备阶段。先检查熔点仪,并拿下散热器,调节显微镜,使日台中心光空完全处于视程中。连接熔点仪热源,打开电源开关并选择手动调节温度至200℃。
(2)将载玻片洗净、吹干后取少量干燥的样品置于载玻片中央,盖上盖玻片,轻轻捻动载玻片使样品均匀的分布在载玻片的中央位置,并确保样品不能太厚,以免影响观察初熔点。待温度升高到140℃左右时,将载玻片放在日台上,微调显微镜使之处在最佳的观察位置,调好后开始密切观察样品变化,当样品边缘开始出现熔化时,迅速记录下此时的温度并记为初熔点。等到样品处于完全熔化状态时,记录下来此时的温度记为终熔点。前后的温度变化值即为产品的熔点范围。
(3)测定完毕后关闭电源并将装置还原至初始状态,并注意清洁卫生。
2.4.2 红外光谱图表征法
红外光谱因其特征性强、操作简便等特点成为有机物结构分析中最常用的方法之一。通过红外光谱图的特定吸收峰可以确定化合物中特定官能团的存在,从而特征的反映出有机化合物的分子结构,并以此来判定化合物是否为目的化合物。其检测步骤如下:
(1)先取一些烘干后的溴化钾放在玛瑙研钵中,再加入产品,加入产品的量为百分之一的溴化钾,然后一起研磨至粉末状。再放入干净的烘干后的表面皿中,之后再放进红外烘箱中烘干。
(2)等到烘干后取出一些样品放入擦拭干净的压片机中压片,压片机的压力应保持在20MPa以上,以确保压片成形,不易损坏。注意压出的片不应该太厚,其透过率保证在40﹪以上时属于较好的片形。
(3)在检测前先要进行系统重置、校准红外分光光度计,并先用纯溴化钾片出图以确保系统的稳定性。再将压好的待测样品的压片放入光束闸中,并将波数扫描开关打开,测量仪器开始扫描,并绘图。
2.5 缓蚀性能评价实验步骤
本实验采用失重法来评价苯并咪唑的缓蚀性能,并根据GB 10124-88金属材料实验室均匀腐蚀全浸试验方法进行实验。实验步骤如下:
(1)准备阶段。选取外形尺寸为50mm×25mm2×mm的标准A3(Q235)钢样若干。经砂纸打磨、去离子水冲洗,然后在无水乙醇溶液中浸泡30min进行脱脂处理,再经热吹风干燥、分析天平称重后备用,并记录下钢样的始重M。
(2)试验溶液的配制。量取125mL的质量分数为36﹪~38﹪的HCl溶液并将其移入250mL的容量瓶中,以配制成浓度约为6moL/L的HCl溶液。然后再分别量取60mL 移入8只烧杯中。之后称取适量的苯并咪唑放入烧杯中,配制成质量浓度范围在
0g/L~7 g/L的苯并咪唑溶液待用。
(3)反应及处理。将处理好的钢样,放入各质量浓度的烧杯中,并用保鲜膜覆盖烧杯口,记录各烧杯中的钢样初始质量M。静置作用72h后,取出。洗干净表面的腐蚀物质后,用热吹风干燥并称重记为M
。
1
(4)腐蚀速率的计算。腐蚀速率的计算公式为
R=8.36×107×(M-M
)/STD
1
其中,R--腐蚀速率,mm/a M--试验前的试样质量,g --试验前的试样质量,g
M
1
S--试样的总面积,cm2
T—试验的时间,h
D—材料的密度,Kg/m 3
3 结果与讨论
本实验在合成阶段是用单因素法去探索苯并咪唑的最佳工艺条件,研究了邻苯二胺与甲酸的摩尔比、催化剂的用量、反应的温度、反应的时间,溶液的pH对产品得率的影响。
3.1 邻苯二胺与甲酸的摩尔比对得率的影响
首先探讨的是在其他条件均不变的情况下,通过变化邻苯二胺与甲酸的摩尔比考察对得率的影响。实验条件及结果列于表3.1中。
表3.1 原料摩尔比对得率的影响
胺酸摩尔比邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度/℃得率/%1:1 5.4 2.3 1.0 2.0 105 22.70
1:2 5.4 4.6 1.0 2.0 105 57.80
1:3 5.4 6.9 1.0 2.0 105 64.92
1:4 5.4 9.2 1.0 2.0 105 74.75
1:5 5.4 11.5 1.0 2.0 105 73.22
1:6 5.4 13.8 1.0 2.0 105 71.18
1:7 5.4 16.1 1.0 2.0 105 69.15
当摩尔比为1:1时,可以看到所得溶液相当粘稠,并且颜色呈棕黄色,且伴有固体颗粒存在,得到的产品即使使用大量的活性炭也无法脱色。可能的原因是在较少的酸存在下,反应不完全,未反应的邻苯二胺被氧化。摩尔比为1:2时的溶液也较黄,产品脱色后微黄。当摩尔比为1:3后,溶液呈现出浅黄色,并且不再需要使用活性炭脱色产品的颜色也都是白色。
通过观察上表可以看出,随着摩尔比的增加,得率先增后减。出现此现象的原因可能是因为该反应属于可逆平衡反应,一开始增加反应物的配比有利于反应向正方向进行,使得率增加。但当加入过量的甲酸后,由于甲酸中含有较多的水,加上反应生成的水,使体系中的水过量反应向逆方向进行,使得得率降低[28]。再者就是因为邻苯二胺上的两个氨基是对称的,酸过量后,有可能反应生成了少量的双酰基化产物,不能环化。使得反应物的得率降低[2]。因此,选择适当过量的酸即胺酸的摩尔比为1:4时是合成苯并咪唑的最佳摩尔配比。
3.2 催化剂种类对得率的影响
本次实验探索比较了磷钨酸与磷酸两种物质作为催化剂的催化效果,条件及结果列于表3.2中。
表3.2 磷钨酸与磷酸催化效果比较
实验序号催化剂/moL 邻苯二胺/g 甲酸/g 得率/%
14 磷钨酸 0.001 5.4 6.9 54.07
15 磷酸 0.001 5.4 6.9 64.24
用磷钨酸作催化剂时,反应结束后溶液呈现出墨绿色,重结晶时有胶状不溶副产物出现。用磷酸催化得到棕色溶液,重结晶时没有胶状不溶副产物出现。从得率看,磷钨酸的催化效果不如磷酸好。熔点测定表明,磷钨酸催化所得产物要比磷酸催化所得产物的熔程范围宽,也表明副产物较多。所以,用磷钨酸作催化剂时,副反应较多,产品得率也较低,综合考虑还是选择磷酸作为本体系催化剂。
3.3 催化剂的用量对得率的影响
在不改变其他条件的情况下,仅仅改变催化剂的用量,来考察催化剂的用量对得率的影响。具体实验条件及结果列于表3.3中。
表3.3 催化剂用量对得率的影响
邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度/℃得率%
5.4
6.9 0.0 2.0 105 61.01
5.4
6.9 1.0 2.0 105 64.92
5.4
6.9 1.5 2.0 105 74.07
5.4
6.9 2.0 2.0 105 7
7.97
5.4
6.9 3.0 2.0 105 76.44
5.4
6.9 4.0 2.0 105 76.05
由上表可知,随着催化剂用量的增加,产品的得率先升后降。在不使用磷酸作催化剂时也有一定的得率,甲酸自身也具有酸性,对反应也有一定催化作用。加入适量磷酸后,反应速率加快,得率也相应提高。但过多的磷酸对本反应体系有害,当其加入量达到4mL时,反应后混合溶液成浅绿色,而不是正常反应时的浅黄色,可以说明有副产物的生成,副反应的增多导致产物得率有所下降。磷酸加入量为2.0mL时,得
率较高,所以定本体系的催化剂的最佳用量为2.0mL。
3.4 反应体系温度对得率的影响
在其他条件均不改变的情况下,仅仅改变反应体系的温度,考察反应体系温度对得率的影响。具体实验条件及结果列于表3.4中。
表3.4 反应体系温度对得率的影响
邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度/℃得率/%
2.7 4.6 2.0 2.0 105 75.08
2.7 4.6 2.0 2.0 110 81.35
2.7 4.6 2.0 2.0 115 8
3.05
2.7 4.6 2.0 2.0 120 78.64
2.7 4.6 2.0 2.0 125 77.29
2.7 4.6 2.0 2.0 135 7
3.56
2.7 4.6 2.0 2.0 145 7
3.22
2.7 4.6 2.0 2.0 155 45.56
上表可知,在105℃~155℃范围内,随着温度的升高,反应所得溶液粘度逐步提高,到155℃时反应体系中出现凝胶状物质,反应后所得溶液稍作冷却即不能从烧瓶中倒出;随着温度的升高,产品得率先升后降,最高可达83.05%,除了155℃外,其余得率均保持在73%~83%的较高范围上。在较低温度下,产物得率随温度升高而增加,是因为该脱水成环反应需要一定的温度,温度升高,有利于传热、传质的进行并可以提高成环,脱水的速率,使产物得率提高。但是当过高的反应温度对有害,当温度达到155℃时产品得率迅速下降,可能是高温下会有较多副产物生成[30];再者就是原料邻苯二胺在高温下容易发生氧化反应,导致参与反应的原料减少,进而导致目标产物收率下降。因此选择反应温度为115℃为反应的最佳温度。
3.5 反应时间对得率的影响
在其他条件均不改变的情况,只改变反应的时间,考察反应时间对得率的影响。具体实验条件及结果列于表3.5中。
表3.5 反应时间对得率的影响
邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度/℃得率%
5.4 5.75 1.0 2.5 105 5
6.61
5.4 5.75 1.0 3.0 105 62.20
5.4 5.75 1.0 3.5 105 67.28
5.4 5.75 1.0 4.0 105 75.25
5.4 5.75 1.0 4.5 105 70.51
5.4 5.75 1.0 5 105 69.83
5.4 5.75 1.0 6 105 52.54
在2.5h~6h范围内随着反应时间延长,反应后所得溶液颜色逐渐加深,由浅黄色变成棕黄色,并且粘度也逐渐增大;随着时间的增加,得率先增后减,但都不低于50%。可能原因是,目标产物是一连串反应的中间产物,随着反应时间的延长,副反应越来越严重,导致4h后产率随时间延长反而下降。有文献[7]报道反应时间延长目标产物深度氧化会加剧,使副产物增多,本体系随反应时间延长体系颜色加深可能正是由于目标产物氧化所致。因此,选择反应时间为4h为反应的最佳的反应时间。
3.6 精制时pH对得率的影响
在合成反应条件均不改变的情况,只改变精制时pH,考察pH对得率的影响。具体实验条件及结果列于表3.6中。
表3.6 精制时pH对得率的影响
邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度℃pH 得率/%
2.7 4.6 2.0 2.0 115 8 71.19
2.7 4.6 2.0 2.0 115 9 74.92
2.7 4.6 2.0 2.0 115 10 82.03
2.7 4.6 2.0 2.0 115 11 74.92
2.7 4.6 2.0 2.0 115 12 68.14
2.7 4.6 2.0 2.0 115 13 68.00
各种pH条件下所得产品均为白色,pH为10时的产品得率最高。原因可能是在此时的苯并咪唑的溶解度最低,所以析出的产物最多。也有文献报道,苯并咪唑溶于酸和强碱,因此选择pH为10时作为反应的最佳pH。
3.7 最佳工艺条件的验证实验
根据系列单因素实验总结出最佳工艺条件,在此条件下进行验证实验。实验条件及结果列于表3.7中。
表3.7 最佳工艺条件下产品的得率
邻苯二胺/g 甲酸/g 磷酸/mL 反应时间/h 反应温度℃得率/%
5.4 9.2 2 4 115 89.60
结果表明,最佳工艺条件下产品得率达89.60%,比系列单因素素实验要高。3.8 产物的表征
3.8.1 产物的物化性质
验证产品是否为目标产物的方法,利用产物的物理化学性质来验证是最基本的方法。如文献报道,苯并咪唑为白色晶体,实验中所得到产品几乎均为白色晶体,又如苯并咪唑微溶与冷水,稍溶于热水。在这个方面,实验中重结晶的过程可以印证这一点。还比如苯并咪唑易溶于酸溶液,在缓蚀实验时将苯并咪唑放入1moL/L的HCl溶液中其立刻溶解也可以证明其是苯并咪唑。因此,通过以上步骤可以初步证明合成的是目的产物苯并咪唑。
3.8.2 熔点表征
选取四个实验产品进行熔点测试,结果见表3.8。
表3.8 产物的熔程范围
产品序号 1 2 3 4
熔程/℃170.0~172.6 170.8~172.7 169.1~173.2 169.4~172.1
文献报道纯品苯并咪唑的熔点为171.0℃,实验所得产物的熔点范围集中在171.0℃左右。考虑到所用的原料邻苯二胺的熔点为103℃~105℃,磷酸的熔点为42.35℃(316K),磷钨酸熔点为~95℃,甲酸本身为液体。可知原料与产物的熔点范围相差较大。可以证明产物是目的产物苯并咪唑。
3.8.3 红外光谱法
为了更充分的证明所得产品即目的产物苯并咪唑,对其作了红外表征,所得图谱如下:
图1 苯并咪唑红外光谱谱图
将所得产品图谱与标准图谱进行比较可以发现其特征峰基本相似。如在1500cm-1,1470cm-1,1362cm-1,有特征吸收峰说明形成了咪唑环,在747cm-1有峰是咪唑环上C-H 的特征峰,在1600cm-1,1580cm-1,1400cm-1上的吸收峰是苯环骨架振动峰,而在3000cm-1附近也出现了苯环上C-H的伸缩振动吸收峰。并且在1620cm-1~1610cm-1处有C=N的吸收峰,在1300cm-1~1250cm-1有C-N的吸收峰[2]。
综上所述,根据产物的物理化学性质、熔点测定、红外光谱表征,可以说明合成出的目的产物是苯并咪唑。
3.9 产物的缓蚀性能初步评价
在应用方面,本论文主要研究了苯并咪唑在缓蚀性能方面的应用。文献报道,苯并咪唑是一种毒性较低,环境友好,缓蚀性能较好,并广泛应用的化合物。
本论文利用失重法和相关规定研究了苯并咪唑的缓蚀性能。实验数据列于表3.9
中。
表3.9 苯并咪唑的缓蚀性能评价数据
试液浓度
g/L 钢样始重
M/g
钢样末重
M1/g
钢样失重/g 腐蚀时间/h 腐蚀速率
10-5mm/a
0 19.1514 17.8008 1.3533 72 7.65
1 19.0285 17.7888 1.2397 7
2 7.00
2 18.9578 17.8588 1.099 72 6.21
3 19.099
4 18.1266 0.9782 72 5.50
4 18.2754 17.4109 0.8654 72 4.89
5 18.5243 17.6915 0.8328 72 4.71
6 19.9390 19.1514 0.7894 72 4.46
7 19.7782 19.0397 07385 72 4.17
实验在平均温度为20℃时进行,腐蚀速率的计算是根据相关公式来计算的。从腐蚀速率可以看出,随着苯并咪唑含量的逐步增加,腐蚀速率在逐渐降低。因为所使用的钢样均为50mm×25mm2×mm的标准件,所以通过钢样的失重也可以验证苯并咪唑是具有一定缓蚀性能的。
结束语
苯并咪唑类化合物在缓蚀、药物应用等方面均具有重要的使用价值,近年来开发利用也很多。鉴于此,本实验利用邻苯二胺与甲酸为原料,以磷酸或磷钨酸为催化剂,探讨出了苯并咪唑合成的最佳工艺条件,即在邻苯二胺与甲酸的摩尔配比为1:4,催化剂为2mL,反应温度为115℃,反应时间为4h,溶液的pH为10时,产物得率可高达89.60﹪。并对产物进行了初步的表征。通过失重法初步评价了苯并咪唑的缓蚀性能。
受实验条件所限,目标产物的表征手段不够齐全,红外光谱仪器尚存在一定的问题。由于时间限制,未能将目标产物与其他物质进行复配,以提高缓释性能。
致谢
论文写到这里已近尾声,同时也在昭示着我向我的大学生涯做最后的道别。有惆怅也有向往,惆怅的是即将面临的分别,惆怅的是再也没有这样无忧无虑的生活,惆怅的是无法归来的青春,更惆怅的是再也没有那样一群纯洁的心灵的交织,师长的,同学的,以及只坐在你旁边一次的那个隔壁班的人……大家彼此真心相待,互助互爱。向往的,往往是美好的,美好的生活,美好的梦想,美好的每一件美好的事情。但更多的美好只有一个宗旨:坚持奋斗,努力拼搏!
完成这篇论文要感谢很多人的帮助,尤其是要感谢我们敬爱的陈冬年老师。他以渊博的知识,严谨的态度,循循善诱的教导方式,引领我在论文完成上走向正确的方向,省却了不必要的麻烦与失误。在此表示我最诚挚的谢意与感激之情!
另外,还要感谢曹鹏老师,张勤老师,邹文杰老师在实验药品与仪器上给予的协助,以及实验过程中给予我帮助的我可爱的同学们!
最后感谢一下这段忙碌的充实的美好的值得回忆的日子!
参考文献
[1] 王箴. 化工辞典[M]. 北京:化学工业出版社,2000.36.
[2] 张景明,戴卫东,王寿武. 2-十三烷基苯并咪唑的合成工艺研究[J].贵州
化工,2009,34(10):11.
[3] 毛郑州,汪朝阳,侯晓娜,等. 苯并咪唑类化合物的合成研究进展[J].有
机化学,2008,28(3):542~547.
[4] 付红蕾,乐长高. 药物中间体2-乙基苯并咪唑的合成[J].华东地质学院学
报,2000,23(4):3473~48.
[5] 王元有. 两种苯并咪唑衍生物的合成方法及其性质[J].扬州工业职业技术
学院学报,2000,23(4):51~54.
[6] 李焱,李俊. 2-取代苯并咪唑合成方法改进[J]. 郑州轻工业学院学报,
2011,26(5):105~106.
[7] 于丽颖,罗亚楠. 2-苯基苯并咪唑的合成[J]. 化学世界,2009,(9):540~
542.
[8] 李莹莹,周永花,郭玉芳等. 苯并咪唑衍生物的合成改进[J]. 有机化学,
2006,26(8):1097~1099.
[9] 陈兴权,赵天生. 2-氨基苯并咪唑的合成[J]. 精细石油化工,2004,(1):
45~46.
[10] 王济奎,张友娟,陈国松,等. 苯并咪唑的光催化一步合成及表征[J].化
学试剂,2000,30(2):115~116+119.
[11] 王济奎,张友娟. 苯并咪唑的光催化一步合成及表征[J].化学试剂,
2008,50(2):68~70.
[12] 刘思全,毕彩丰,王立国. 2-甲胺基苯并咪唑的合成及表征[J].精细化
工,2009,26(1):81+103~104.
[13] 刘长令,翟煜翥,张云晓,等. 防治灰霉病用杀菌剂的开发[J].农药,
2000,39(3):1~6.
[14] 田敏,王璐瑶,陈邦,等. 芳香基苯并咪唑衍生物的合成、表征及抑菌活
性研究[J].化学通报,2005,68(9):709~713.
[15] 王敏,张一宾. 聚咪唑基杀菌剂的研究开发进展[J].现代农药,2003,2
(1):36~38.
[16] 高成庄,李焱. 苯并咪唑衍生物的特性及应用研究进展[J].郑州轻工业学
院学报(自然科学版),2007,22(2/3):35~36.
[17] 高学军,李庆章. 苯并咪唑氨基甲酸酯类抗蠕虫药物作用机理研究进展
[J].东北农业大学学报,2004,35(4):492~495.
[18] 李焱,马会强,王玉炉. 苯并咪唑及其衍生物合成与应用研究进展[J].有
机化学,2008,28(2):210~217.
[19] 温玉麟. 药物与化学物质毒性数据[M].天津:天津科学技术出版社,
1989.55.
[2O] 胡莲跃,张胜涛,吴永英等. 苯并咪唑对黄铜缓蚀性能及吸附行为的影响[J].腐蚀科学与防护技术,2011,23(4):339~341.
[21] 史志龙,庞正志. 新型铜酸洗缓蚀剂烷基苯并咪唑的研究[J].北京化工大
学学报,2002,29(2):52~54.
[22] 王清华,杨官汉. 苯并咪唑衍生物抗腐蚀机理的研究[J].润滑与密封,
2002,29(2):52.
[23] 胡建春,李春志,严奇,等. 苯并咪唑及其衍生物的缓蚀性能[J].材料保
护,2011,44(11):75~78.
[24] 沈建,余鼎声,庞正智. 苯并咪唑类化合物作为碳钢缓蚀剂的研究[J].北
京化工大学学报,2005,4(32):110~112.
[25] 胡莲跃,张胜涛,黄小红,等. 苯并咪唑在KOH溶液中对锌材的缓蚀性能
[J].材料保护,2011,44(8):14~17.
[26] GB 10124-88金属材料实验室均匀腐蚀全浸试验方法[S] .
[27] 彭兴华. 大学化学实验2合成实验与技[M].北京:化学工业出版社,
2007.8.
[28] 史子兴,庞正志. 2-烷基苯并咪唑的合成及结构表征[J].北京化工大学学
报,1997,24(2):27~32.
[29] 常慧,尹浩坚,宁满侠. 微波催化快速合成苯并咪唑及其2-烷基衍生物[J].
东莞理工学院学报,2008,15(3):88~90.
[30] 唐欣,唐有根,刘小平,等. 2-取代基苯并咪唑合成工艺改进[J].广州化
学,2008,33(4):38~41.
万方数据
万方数据
过氧化氢氧化苯甲醛合成苯甲酸的研究 作者:冯冬然, 张光霞, 殷利河, 边延江 作者单位:廊坊师范学院,河北,廊坊,065000 刊名: 廊坊师范学院学报(自然科学版) 英文刊名:JOURNAL OF LANGFANG TEACHERS COLLEGE 年,卷(期):2008,8(6) 本文读者也读过(10条) 1.于丽颖.吉慧杰.王悦虹.YU Li-ying.JI Hui-jie.WANG Yue-hong过氧化氢氧化苯甲醛合成苯甲酸[期刊论文]-辽宁化工2008,37(12) 2.李贵贤.张烨红.毛志红.范宗良.谭学苓.汪孔照.LI Gui-xian.ZHANG Ye-hong.MAO Zhi-hong.FAN Zong-liang. TAN Xue-ling.WANG Kong-zhao磷钼杂多酸季铵盐催化氧化合成苯甲酸[期刊论文]-化学与生物工程2009,26(7) 3.吴旭.谢鲜梅.WU Xu.XIE Xian-mei乙酸存在下钴铝类水滑石催化氧化苯甲醛合成苯甲酸[期刊论文]-太原理工大学学报2010,41(2) 4.谢鲜梅.吴旭.杜亚丽.严凯.王志忠.XIE Xian-mei.WU Xu.DU Ya-li.YAN Kai.WANG Zhi-zhong镍铝类水滑石在乙酸介质下催化氧化苯甲醛合成苯甲酸[期刊论文]-现代化工2006,26(z2) 5.江秀清.林海昕.林敏.JIANG Xiu-qing.LIN Hai-xin.LIN Min微波辐射相转移催化法合成苯甲酸[期刊论文]-厦门大学学报(自然科学版)2008,47(z2) 6.丁元生.罗志臣.周端文.Ding Yuansheng.Luo Zhichen.Zhou Duanwen Keggin型配合物 [(CH2)5NH2]4SiMo12O40的合成及其催化氧化合成苯甲酸的研究[期刊论文]-精细石油化工2010,27(3) 7.刘春生.严红燕.张少华.罗根祥清洁催化氧化苯甲醛制备苯甲酸[期刊论文]-辽宁石油化工大学学报2004,24(2) 8.张静波.张贵荣.钮东方.陆嘉星银电极电催化溴苯与C02合成苯甲酸[会议论文]-2008 9.严红燕.程云.刘春生.姜恒.罗根祥甲烷磺酸铜催化氧化苯甲醛制苯甲酸[期刊论文]-抚顺石油学院学报2003,23(3) 10.谢鲜梅.吴旭.杜亚丽.严凯.王志忠镍铝类水滑石在乙酸介质下催化氧化苯甲醛合成苯甲酸[会议论文]-2006 引用本文格式:冯冬然.张光霞.殷利河.边延江过氧化氢氧化苯甲醛合成苯甲酸的研究[期刊论文]-廊坊师范学院学报(自然科学版) 2008(6)
毕业设计(论文)设计题目苯甲酸的合成工艺 办学学院扬州工业职业技术学院 专业应用化工 姓名李功进 起讫日期2015-3-1 指导教师李淑丽 2015 年 3 月 1 日
摘要苯甲醛因有广泛的用途,年需求量较大、市场前景好和经济效益高的特点。近年来涌现出许多新颖性的方法制备苯甲酸。本文根据反应原料的不同进行了归纳总结。 关键词苯甲醛;合成;KMnO4;甲苯 一、苯甲酸的概述 (一)苯甲酸分子结构的分析 苯甲酸又称安息香酸,分子式为C6H5COOH,羧基直接与苯环碳原子相连接的最简单的芳香酸,是苯环上的一个氢被羧基(-COOH)取代形成的化合物。为无色、无味片状晶体。熔点122.13℃,沸点249℃,相对密度1.2659(15/4℃)。在100℃时迅速升华,它的蒸气有很强的刺激性,吸入后易引起咳嗽。微溶于水,易溶于乙醇、乙醚、氯仿、苯、甲苯、二硫化碳、四氯化碳和松节油等有机溶剂。 苯甲酸是弱酸,比脂肪酸强。它们的化学性质相似,都能形成盐、酯、酰卤、酰胺、酸酐等,都不易被氧化。苯甲酸的苯环上可发生亲电取代反应,主要得到间位取代产。苯甲酸一般常作为药物或防腐剂使用,有抑制真菌、细菌、霉菌生长的作用,药用时通常涂在皮肤上,用以治疗癣类的皮肤疾病。用于合成纤维、树脂、涂料、橡胶、烟草工业。最初苯甲酸是由安息香胶干馏或碱水水解制得,也可由马尿酸水解制得。工业上苯甲酸是在钴、锰等催化剂存在下用空气氧化甲苯制得;或由邻苯二甲酸酐水解脱羧制得。苯甲酸及其钠盐可用作乳胶、牙膏、果酱或其他食品的抑菌剂,也可作染色和印色的媒染剂[1] 苯甲酸是化学工业,尤其是石油化学工业中重要的有机原料和产品之一,它广泛用于生产医药中间体、食品添加剂、化妆品及化工产品,如苯酚、己内酰胺的工业生产中。全世界苯甲酸产量在200 万吨/年以上,仅制造苯酚和己内酰胺就消耗苯甲酸80 万吨/年以上。苯甲酸及其钠盐、钾盐均可作为酸性食品防腐剂,目前其消费量居我国防腐剂用量之首。 (二)苯甲酸物理性质 表1-1列出了苯甲酸的一些物理性质。
苯甲醇与苯甲酸的制备实验 一、实验原理 利用坎尼扎罗(Cannizzaro)反应由苯甲醛制备苯甲醇和苯甲酸。 坎尼扎罗反应是指无α-活泼氢的醛类在浓的强NaOH 或 KOH 水或醇溶液作用下发生的 歧化反应。此反应的特征是醛自身同时发生氧化及还原作用,一分子醛被氧化成羧酸(在碱性溶液中成为羧酸酸盐),另一分子醛则被还原成醇。 主反应: 机理:醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以 氢负离子的形式转移到另一分子的羰基不能碳原子上。 二、反应试剂、产物、副产物的物理常数 三、药品
四、实验流程图 五、实验装置图 图 1 磁力搅拌器图2分液漏斗的振摇方法图3分液漏斗图4抽滤装置
六、实验内容 往锥形瓶中加12.0g(0.21mol) 氢氧化钾和12ml 水,放在磁力搅拌器上搅拌,使氢氧化钾溶 解并冷至室温。在搅拌的同时分批加入新蒸过的苯甲醛,每次加入 2-3ml,共加入14g,0.13mol) 。加后应塞紧瓶口,若锥形瓶内温度过高,需适时冷却。继续搅拌 13.5ml( 约60min,最 后反应混合物变成白色蜡糊状。 (1) 苯甲醇 向反应瓶中加入大约 45ml 水,使反应混合物中的苯甲酸盐溶解,乙 醚分三次萃取苯甲醇,合并乙醚萃取液。保存水溶液留用。 转移至分液漏斗中,用 45ml 依次用15ml25% 亚硫酸氢钠溶液及8ml 水洗涤乙醚溶液,用无水硫酸镁干燥。水浴蒸去乙 醚后,继续蒸馏,收集产品,沸程204-206℃,产率为75%。 纯苯甲醇有苦杏仁味的无色透明液体。沸点bp=205.4 ℃,折光率 =1.5463。 (2)苯甲酸 在不断搅拌下,往留下的水溶液中加入浓盐酸酸化,加入的酸量以能使刚果红试纸由红变蓝 为宜。充分冷却抽滤,得粗产物。 粗产物用水重结晶后晾干,产率可达80%。 纯苯甲酸为白色片状或针状晶体。熔点 mp=122.4 ℃。 (一)制备阶段 1.准备锥形瓶:一只100ml锥形瓶。 2.加药品与歧化反应:向锥形瓶中加12.0g氢氧化钾和12ml水,向瓶内放入一只搅拌子,然后将 锥形瓶放在磁力搅拌器上搅拌,使氢氧化钾溶解并冷至室温。在搅拌的同时分批加入新蒸过的苯 甲醛,每次加入 2-3ml ,共加入 13.5ml( 约 14g, 0.13mol) 。加后应塞紧瓶口,若 锥形瓶内温度过高,需适时冷却。继续搅拌60min ,最后反应混合物变成白色蜡糊状。 【为避免歧化反应过快产生大量热量,造成温度过高增加氧化副反应,故需将苯甲醛分几批加 入】 (二)后处理阶段 1.分离苯甲醇 (1)加水溶解:向反应瓶中加入大约45ml水,使反应混合物中的苯甲酸盐溶解,转移至分液 漏斗中。 【在磁力搅拌器上尽量搅拌时间长一些,以保障苯甲酸钾盐充分溶解在水中,减少与苯甲 醇分子的包裹,有利于下一步的乙醚萃取】 (2)乙醚萃取:用45ml乙醚分三次萃取苯甲醇,合并乙醚萃取液。保存水溶液留用(含有苯甲 酸钠盐)。 【每次萃取振荡时间不能过长,每振荡2-3 次,就要进行放气一次,如此重复2-3 次即可。避免漏斗内产生大量乙醚气体而喷出。】 (3)亚硫酸氢钠洗涤:用15ml25%亚硫酸氢钠溶液洗涤乙醚溶液,洗涤除去未反应的苯甲 醛。 【洗涤时振荡不能长时间振荡,避免下层的水中溶解过多的乙醚而降低亚硫酸氢钠在水中 的溶解度,可能达到饱和析出大量晶体。】 (4)水洗涤:8ml水洗涤乙醚溶液,除去上一步洗涤后残留的亚硫酸氢钠。 【因下一步紧接着的操作是干燥粗产品,所以水洗涤后应该多静止几分钟,再分去下层的 水层。】
苯甲醇和苯甲酸的制备 一.实验目的 1.学习由苯甲醛制备苯甲醇和苯甲酸的原理和方法 2.巩固机械搅拌器的使用 3.进一步掌握萃取、洗涤、蒸馏、干燥和重结晶等基本操作 4.学会低沸点溶剂的处理 二.实验原理 无α-H的醛在浓碱溶液作用下发生歧化反应,一分子醛被氧化成羧酸,另一分子醛则被还原成醇,此反应称坎尼扎罗反应。本实验采用苯甲醛在浓氢氧化钠溶液中发生坎尼扎罗反应,制备苯甲醇和苯甲酸。反应物加水溶解后,用乙醚加以萃取,乙醚层经洗涤、干燥、蒸馏,得到苯甲醇;水层经酸化得到苯甲酸。反应式如下: CHO 2+NaOH CH2OH + COONa COONa +HCl COOH+NaCl 苯甲醛如果长时间放置,会被空气氧化,因此存在以下的副反应:CHO +O 2 COOH 三.实验准备 1.主要试剂及仪器 1)试剂
苯甲醛10 mL (mol),氢氧化钠8g ,浓盐酸,乙醚,饱和亚硫酸氢钠溶液,10%碳酸钠溶液,无水硫酸镁。 2)仪器 100mL 圆底烧瓶,球形冷凝管,分液漏斗,直形冷凝管,蒸馏头,温度计套管,温度计(250℃),支管接引管,锥形瓶,空心塞,量筒,烧杯,布氏漏斗,吸滤瓶,表面皿,红外灯,机械搅拌器。 2.主要物料物理常数 四.实验装置及操作要点 制备苯甲醇和苯甲酸采用回流搅拌装置,实验装置如图1所示。乙醚沸点低,要注意安全。蒸馏低沸点液体的装置如图2所示。
图1.制备苯甲醇和苯甲酸的反应装置 图2.蒸乙醚的装置图 【干燥操作要点】 1.干燥剂不是越多越好 2.干燥操作在干燥的带有塞子的锥形瓶中进行。加入干燥剂后,加塞旋摇,放置一段时间,根据干燥情况补加。 3.干燥彻底,液体透明澄清,干燥剂棱角分明、不粘结一团、不粘在瓶壁上。 【蒸馏乙醚的操作要点】 1.在实验室使用或者蒸馏乙醚时,实验台附近严禁有明火。因为乙醚容易挥发,且易燃烧,与空气混合到一定比例时极易发生爆炸。 2.蒸乙醚时可在接引管支管上连接一长橡皮管通入水槽的下水管内或引入室外。 3.接收器用冷水浴冷却。
一种苯甲醛的制备方法 一、技术领域: 本发明是一种生产广泛用于医药、染料、香料等有机合成重要中间体苯甲醛的生产新工艺。 二、背景技术及存在技术问题 现有苯甲醛生产技术普遍采用商品甲苯经沉降分离脱水后的甲苯和氯气在搪瓷反应釜内在普通光源的催化下,通过间歇式氯化反应制取苄叉二氯和苄基氯混合物,再通过间歇式分馏处理,得85%左右的苄叉二氯用纯碱溶液水解制的苯甲醛。 该工艺的主要缺点是: 1、该反应对铁杂质要求较高,瓷非常容易脱落,一旦搪瓷脱落,反应釜 会很快穿孔报废,同时由于搪瓷脱落后料液和铁之间接触后,苄基氯 会反应剧烈的聚会反应,有爆炸的危险。 2、传统工艺采用是通过静臵的方式粗略精致甲苯,这样处理过的甲苯水 含量通常在300ppm以上,这样在反应是会形成盐酸溶液,对设备产生 腐蚀,同时在金属酸溶液存在下,甲苯会发生苯环上氯代反应,不仅 降低了苄叉二氯的收率,而且形成难以分离的环氯副产物。 3、传统工艺采用普通全波段的光源,不仅选择性不好,氯的吸收效率也 较低) 4、传统方式间歇式分馏方式,不仅生产效率极低,而且分馏效果也比较 差,苄叉二氯中的苄基氯的含量在15%左右 5、由于苄基氯和苯甲醛的沸点几乎相同仅相差0.4度,采用传统的分馏 操作,很难使二者分离,传统工艺是通过苄基氯在12%的碳酸钠水溶 液中水解成苄醇,再通过分馏的方法提纯苯甲醛,这样,苄基氯就通 过纯碱转化成苄醇成为分馏后釜残废液了,不仅降低了苯甲醛的收率, 而且增加了污染。 三、技术方案 经过共沸脱水后的精致甲苯和氯气在特殊材质的反应器内,通过高压紫外灯(波长为400nm左右)轴向光照在95-110度之间反应生成苄基氯、苄叉二氯
氯代苯甲醛的应用及合成工艺 (江苏常州江东化工有限公司费平213014) The application and synthesis process of chloro-phenylaldehyde 1.前言 氯代苯甲醛的种类较多,主要是指以下几种,它们的合成方法有许多相似之处:Cl Cl Cl CHO CHO Cl—CHO CHO Cl Cl 邻氯苯甲醛对氯苯甲醛2,4—二氯苯甲醛2,6—二氯苯甲醛氯代苯甲醛在一些工业发达国家,早已形成了规模化的工业生产,工艺上也居于领先地位。我国氯代苯甲醛的开发起步较晚,但近几年随着市场需求量的增加和原料氯代甲苯质量的提高,我国氯代苯甲醛的研究开发工作也有了长足的发展,并逐步形成了工业化生产,工艺技术不断改进,质量不断提高,除满足国内市场的需求外,还大量出口创汇,取得了较好的经济和社会效益。 2.用途 氯代苯甲醛都是高附加值的精细化工产品,是农药、医药、染料、香料、颜料及其它有机合成的重要中间体。 目前国内生产应用最多的主要是:邻(对)氯苯甲醛,这里就仅介绍它们的工业应用。 邻氯苯甲醛的应用:邻氯苯甲醛与丙酮发生Claisen-Schmidt反应生成的双烯—酮衍生物,在医药上具有消炎作用;与盐酸羟胺缩合生成的邻氯苯甲肟,是合成药物的重要中间体;在农业上可用以合成除草剂、植物生长调节剂,高效杀螨剂螨死净;染料上用以制造邻磺基苯甲醛钠及荧光增白剂;某些记录材料的中间体。 对氯苯甲醛的应用:对氯苯甲醛在医药上用以合成芬那露,氨基氨络酸等;染料上用以制备酸性蓝(C.I.,酸性蓝83,90,100,109等);农业上用以生产新型除草剂麦敌散,植
绿色氧化苯甲醇合成苯甲醛研究进展
绿色氧化苯甲醇合成苯甲醛研究进展 摘要:苯甲醛是一种在有机反应中重要的有机合成中间体,其在农药、染料、香料、化妆品和制药等工业中被大量的应用。苯甲醇选择性催化氧化合成苯甲醛的反应,无论是在工业方面还是在有机化学理论方面,都有着很重要的作用。本论文从苯甲醇和苯甲醛性质和用途写起,主要写了杂多酸催化,金属催化,离子液体催化和负载型过渡金属有机络合物催化的研究进展。 关键词:绿色氧化苯甲醇苯甲醛催化 苯甲醇合成苯甲醛是有机化学基本反应之一,苯甲醛工业生产和有机反应中被大量应用。苯甲醇合成苯甲醛的反应可达到最高产率,其氧化在工业上和化学研究方面受到广泛注意[1]。传统将醇氧化成醛的合成方法是在有机体系中用锰、铬的化合物做氧化剂,此方法不仅会产生大量污染环境的副产物和废物,腐蚀设备,而且产品分离困难产率较低。因此,研究新的催化剂,探索选择性合成苯甲醛的氧化催化体系,这已经是当今一个刻不容缓的课题[2]。 1苯甲醇、苯甲醛 1.1苯甲醛 苯甲醛是有机化合物中应用非常广的芳香醛,也是最简单的最基本的醛类化合物之一。颜色为无色或浅黄色,是一种具有强折射率的容易挥发的油状液体,味道为苦杏仁味,燃烧时有香味。苯甲醛在常温下微溶于水,能够混溶在乙醇、乙醚等有机溶剂中。苯甲醛在制造颜料方面是一种非常重要的中间产物,日常生活中,在制造香料、调味剂和药品方面有着普遍应用,但其在无氯的条件下才可以安全的使用。即使用作工艺品,苯甲醇的含量也必须高于98%,其他物质的含量必须低于2%。苯甲醛总产量的60%-80%应用在日常生活所用的香料和药品中[3]。 1.2苯甲醇 苯甲醇是一种在有机化合物中最简单和最基本的醇类化合物之一。自然界中的苯甲醇在香油精中以酯的形式大量存在。苯甲醇有很微弱的香味,常被用作香料的稀释剂。在常温下苯甲醇易溶于有机溶剂,能溶于水。苯甲醇的饱和溶液配置为将1 g苯甲醇溶于25ml 水中。苯甲醇除了被氧化成苯甲醛外,也容易被氧化成苯甲酸,例如用硝酸做氧化剂氧化,因浓度和温度的变化,其产物也在发生变化,醛或酸所占总产物的比例也不同。 2杂多酸催化剂 杂多酸催化剂在化学反应中既表现出良好的酸性,又表现出良好的氧化性能。在苯甲醇合成苯甲醛催化方面有着很好的应用前景,是一种优秀的催化剂[4]。 2.1磷钨杂多酸 磷钨酸在醇氧化成醛的相关化学反应中表现出很好的催化性能,不同类型的氧化剂对该反应的影响效果不同[5]。经研究表明,在温度,压强等物理因素相同的条件下,使用能够在表面活性表现良好的氧化剂和使用非表面活性的氧化剂时,前者反应活性更高,反应过程中的搅拌转速也会影响着反应速率,两次对比实验也要确保转速的相同。例如,过氧化氢是一种表面活性良好的氧化剂,磷钨酸、双氧水和水组成的体系以磷钨酸作为催化剂,表现出良好的催化性能,该反应使用的都是绿色试剂,而且磷钨酸的用量少,很大程度上减短了反应时间,反应过程中易于操作,最终获得的苯甲醛产率高,以上说明该体系是一种高产率的绿色催化体系,具有广泛的应用范围[6-7]。 2.2 Dawson型磷钼钒杂多酸盐
综述:苯甲酸的合成方法 摘要:本文从催化剂的分类来介绍苯甲酸的合成方法,总共有两大类方法:一类是有机催化剂催化有机物合成苯甲酸,另一类是无机催化剂催化有机物合成苯甲酸。 关键字:苯甲酸的合成方法 有机催化剂 无机催化剂 0.前言 苯甲酸是一种重要的化工原料,主要用于生产苯甲酸钠食品防腐剂、染料中 间体、农药、增塑剂、医药、香料等,还可用作钢铁设备的防锈剂。苯甲酸可以由甲苯的氧化,苯甲醇的氧化,苯甲醛的氧化制备。工业上苯甲酸的合成大致可以分为两类:一类是用有机催化剂催化有机物合成,另一类是用无机催化剂催化有机物合成。 1.有机催化剂催化 对于甲苯氧化制备苯甲酸,李涛等人以不同钴基吡唑配合物为催化剂进行甲 苯氧气液相氧化反应,发现)5,3(,'二甲基吡唑二--N N 溴化钴在甲苯氧化反应中具有较好的催化反应活性,比乙酸钴催化性能更好。[1] 对于苯甲醇,孙晓云等人用甲基三辛基氯化铵和钨酸钠一步法合成甲基三辛 基季铵钨酸盐离子液体112231783])()[(O W H C n N CH -,以该离子液体为催化剂,在无反应溶剂条件下催化过氧化氢氧化苯甲醇生成苯甲酸,并且确定优化条件:反应温度70℃,苯甲醇用量5 mmol ,催化剂用量是底物的0.4%(摩尔分数),30%过氧化氢用量2 mL ,苯甲醇的转化率可达99%,苯甲酸选择性为98%。[2]郭飞燕等人以质量分数为30%的22O H 作氧化剂,磷钨酸及磷钨酸盐作催化剂催化合成苯甲酸[3] 对于苯甲醛,李贵贤等人采用四丁基溴化铵和磷钼酸反应合成了一系列 Keggin 型磷钼酸季铵盐催化剂,傅立叶红外光谱分析表明,所制备的催化荆具有典型的杂多阴离子结构。将其应用于冰乙酸作溶剂、30%过氧化氢作氧化刺催化氧化苯甲醛制苯甲酸的反应过程中,结果表明,催化荆中各组分在双氧水作用下催化活性较高,任一组分单独作用时催化活性较低,只有当催化剂中季铵盐与磷钼酸配比合适时,催化效果才会最优,其中40122494])[(O PMo H N H C 的催化活性
Cannizzaro反应—苯甲醇和苯甲酸 一、实验目的 1、掌握苯甲醇和苯甲酸的制备; 2、学习Cannizzaro(康尼扎罗)反应原理; 3、熟练掌握实验中的萃取、洗涤等基本操作。 二、实验原理 1、苯甲酸和苯甲醇的相关性质 2、Cannizzaro反应及反应机理 没有α-氢的醛,如苯甲醛,在强碱作用下,会发生分子间的氧化还原反应,一分子被还原成苯甲醇,另一分子被氧化成苯甲酸,即Cannizzaro反应。
反应机理: 3、本实验的相关反应 主反应: 副反应: 4、刚果红试纸 刚果红试纸是由刚果红溶液浸泡制成。刚果红呈枣红色粉末状,能溶于水和酒精,遇酸呈蓝色。它不仅能作染料,也用作指示剂。刚果红是酸性指示剂,变色范围为3.5到5.2,碱态为红色,酸态为蓝紫色。有人认为刚果红毒性很大,其实是错误的。刚果红能做为药剂成分.
三、实验装置示意图 四、实验步骤 1、加料,歧化反应:100ml锥形瓶中,加18gKOH ,18ml 水和10ml苯甲醛,该反应是 两相反应,不断振摇是关键。得白色糊状物。静置 2、萃取,分离:加水溶解,置于分液漏斗中。每次用20ml乙醚萃取,共萃取水层3次(萃
取苯甲醇),水层保留。 3、洗涤醚层:依次用亚硫酸氢钠(饱和)、10%碳酸钠、水各5ml洗涤醚层。除去苯甲醛, 酸性亚硫酸氢钠盐 4、干燥,蒸馏:用无水硫酸镁干燥半小时。水浴回收乙醚。用空气冷凝管收集苯甲醇 200-204℃馏分。N201.5396,约4-5g. 5、酸化,重结晶:浓盐酸酸化使刚果红试纸变蓝,冷却析出苯甲酸。必要时用水重结晶。.mp.121-122℃ 6、测折射率 五、注意事项 1.如果第一步反应不能充分振摇,会影响后续反应的产率。如混合充分,放置24小时后混合物通常在瓶内固化,苯甲醛气味消失; 2.用分液漏斗分液时,水层从下面分出,乙醚层要从上面倒出,否则会影响后面的操作;3.用干燥剂干燥时,一定要澄清后才能倒在蒸馏瓶中蒸馏,否则蒸出的产物不纯; 4.水层如果酸化不完全,会使苯甲酸不能充分析出,导致产物损失。
学生姓名:小田田学号:专业班级: 实验类型:□验证■综合□设计□创新实验日期:2013年4月24日实验地点: 同组学生姓名:指导教师:实验成绩: 实验八:苯甲醇和苯甲酸的制备 一、实验目的 1.学习由苯甲醛制备苯甲醇和苯甲酸的原理和方法。 3.进一步掌握萃取、洗涤、蒸馏和干燥等基本操作。 二、产品介绍 苯甲醇是最简单的芳香醇之一,可看作是苯基取代的甲醇。在自然界中多数以酯的形式存在于香精油中。中文别名苄醇;。苄醇是极有用的定香剂,用于配制香皂;日用化妆香精。但苄醇能缓慢地自然氧化,一部分生成苯甲醛和苄醚,使市售产品常带有杏仁香味,故不宜久贮。苄醇在工业化学品生产中用途广泛;医药;合成树脂溶剂;可用作尼龙丝;纤维及塑料薄膜的干燥剂,染料;纤维素酯;酪蛋白的溶剂,制取苄基酯或醚的中间体。同时,广泛用于制笔(圆珠笔油);油漆溶剂等。 苯甲酸为具有苯或甲醛的气味的鳞片状或针状结晶,具有苯或甲醛的臭味。在100℃时迅速升华,它的蒸气有很强的刺激性,吸入后易引起咳嗽。微溶于水,易溶于乙醇、乙醚等有机溶剂。苯甲酸是弱酸,比脂肪酸强。它们的化学性质相似,都能形成盐、酯、酰卤、酰胺、酸酐等,都不易被氧化。苯甲酸的苯环上可发生亲电取代反应,主要得到间位取代产。 三:反应式 主反应: CHO + NaOH 2 CH2OH + COONa COONa + HCl COOH + NaCl 副反应:CHO COOH +O2
学生姓名:小田田学号:专业班级: 实验类型:□验证■综合□设计□创新实验日期:2013年4月24日实验地点: 同组学生姓名:指导教师:实验成绩: 四:主要试剂及产品的物理常数 五:试剂与仪器 1、主要仪器 : 125mL 圆底烧瓶、分液漏斗,蒸馏装置、烧杯等。 2、主要试剂规格及用量:
开题报告题目苯甲醛的合成工艺技术 姓名 所在系部 专业班级 指导教师 2012 年 12 月
一、选题的背景与研究意义 背景:苯甲醛(Benzaldehyde),无色或浅黄色,是一种强折射率的挥发性油状液体,具有苦杏仁味,故又称苦杏仁油。 苯甲醛的熔点-26℃,沸点178℃,相对密度 1.0415(104℃)。能与乙醇、乙醚、氯仿等混溶,微溶于水。能进行水蒸气蒸馏。苯甲醛的化学性质与脂肪醛类似,但也有不同。苯甲醛不能还原费林试剂;用还原脂肪醛时所用的试剂还原苯甲醛时,除主要产物苯甲醇外,还产生一些四取代邻二醇类化合物和均二苯基乙二醇。在氰化钾存在下,两分子苯甲醛通过授受氢原子生成安息香。苯甲醛还可进行芳核上的亲电取代反应,主要生成间位取代产物,例如硝化时主要产物为间硝基苯甲醛。 研究意义:本论文对甲苯侧链氯化水解法制备苯甲醛产品的方法进行了研究,提高了苯甲醛的收率,并得到了最佳工艺条件。重点考察了直接碱性水解和酸碱复合水解,甲苯氯化时反应时间、光照效率,蒸馏时真空度、塔顶出料液时的温度对收率的影响。从国内市场来看,无氯苯甲醛市场需求很大。为了满足市场对苯甲醛日益增长的需要,研究和开发一条经济、有效的制备苯甲醛的新途径是十分必要和有意义的。甲苯氯化水解法就是一种适于大量生产无氯苯甲醛的好方法。 ①可以充分利用甲苯催化重整过程的迅速发展,使得甲苯价廉易得,产量不断增长。从甲苯的有效利用和经济效益两方面来考虑,甲苯作为苯甲醛的生产原料,可说是最理想的选择。 ②生产大量无氯苯甲醛,以满足食品、香料、化妆品、医药等工业对无氯苯甲醛的日益增长的需要。 ③由甲苯生产无氧苯甲醛可创造可观的经济利益。甲苯的价格较苯甲醛低得多,甲苯一般为4000元/吨,而苯甲醛为16000元/吨。 总之甲苯氯化水解法是一种价廉物美的苯甲醛生产方法,生产能力大,污染少,是值得研究开发的方法。 由于甲苯液相空气氧化法仍存在低选择性或低收率的最大不足,所以至今仍没找到一种有效的适合工业生产的方法。其根本原因是苯氧化成苯甲醛,从而很难控制氧化深度。 甲醛极易被O 2
苯甲酸的制备实验 一、实验原理 氧化反应是制备羧酸的常用方法。芳香族羧酸通常用氧化含有α-H的芳香烃的方法来制 备。芳香烃的苯环比较稳定,难于氧化,而环上的支链不论长短,在强裂氧化时,最终都氧化成羧基。 制备羧酸采用的都是比较强烈的氧化条件,而氧化反应一般都是放热反应,所以控制反应在一定的温度下进行是非常重要的。如果反应失控,不但要破坏产物,使产率降低,有时还有发生爆炸的危险。 主反应: 二、反应试剂、产物、副产物的物理常数 三、药品 四、实验装置图
图1 电动搅拌器图2回流搅拌装置图3抽滤装置 五、实验流程图 六、实验内容 在安装有电动搅拌器、回流冷凝管的250ml三口圆底烧瓶中放入1.4ml甲苯和70ml水,加热至沸。从冷凝管上口分批加入4.3g高锰酸钾;粘附在冷凝管内壁的高锰酸钾最后用25ml 水冲洗入瓶内。继续煮沸并间歇摇动烧瓶,直到甲苯层几乎近于消失、回流液不再出现油珠(约需4-5h)。 将反应混合物趁热减压过滤,用少量热水洗涤滤渣(MnO2)。合并滤液和洗涤液,于冰水浴中冷却,然后用浓盐酸酸化(刚果红试纸检验),至苯甲酸析出完全。将析出的苯甲酸减压过滤,用少量冷水洗涤,挤压去水分。把制得的苯甲酸放在沸水浴上干燥。产量:约1.0g。若要得到纯净产品,可在水中进行重结晶。 纯净的苯甲酸为白色片状或针状晶体。熔点mp=122.4℃。 (一)制备过程
1.安装制备装置:如图(1)(2),首先放置好电动搅拌器,然后由下往上安装各个仪器,即将控温电热套平放在桌面上,接着固定250ml三口圆底烧瓶(瓶底不能接触电热套),安装搅拌棒(要保证搅拌棒转动时不能接触瓶底)、并将搅拌棒与电动搅拌器电机连接固定、调节(用手转动搅拌棒观察有无摩擦现象,若有摩擦,需调整消除),一侧口连接回流冷凝管(万用夹夹在冷凝管的中部;冷凝管的上口应该是敞口的,不能用塞子),另一侧口安装温度计(水银球要插到液面以下)。 2.加药品:从连有温度计的侧口,依次加入1.4ml甲苯、70ml水和4.3g高锰酸钾(或加药品顺序为4.3g高锰酸钾、100ml水和1.4ml甲苯)(一次性加入高锰酸钾即可)。 3.加热:先打开电动搅拌器电源开关,慢慢旋转调速旋钮使电动搅拌器的搅拌棒逐渐转起来,由小变大,正常搅拌的时候,再开始加热,直至微微沸腾。控制加热速度,使蒸气体不超过冷凝管下面数第二个球部为宜,直到甲苯层几乎近于消失、回流液不再出现油珠(约1-2h)。【注:因氧化反应是放热反应,故在制备反应的整个过程中,要保证电动搅拌器不能停止,否则可能会发生反应液喷出的现象。一旦出现故障需要调节搅拌器的话,必须先撤去电热套,同时用手转动搅拌棒进行搅拌才行。】 (二)后处理过程 1.加亚硫酸氢钠:因氧化剂高锰酸钾是过量的,反应完后反应液仍呈紫色,可从冷凝管上口分次加入少量饱和亚硫酸氢钠溶液,直到使反应液紫色褪去为止。(除去过量的高锰酸钾)【注:操作仍在上面的搅拌装置中进行,这时可以停止加热,撤去电热套,但搅拌不能停。在搅拌的同时,慢慢地从冷凝管上口分批加入饱和亚硫酸氢钠溶液,以防止带入大量空气气体而引起爆沸、喷出反应液。饱和亚硫酸氢钠溶液浓度为40%】 【在本实验中,亚硫酸氢钠的最小用量为与过量的高锰酸钾的mol量相当,即0.001mol,为0.10g;最大用量为与4.3g(0.027mol)高锰酸钾的mol量相当,即2.81g,故亚硫酸氢钠的用量范围为0.10-2.81g。亚硫酸氢钠用量不要过量太多,否则在后面的酸化时会与盐酸作用产生太多的亚硫酸而再分解为二氧化硫气体。】 2.趁热减压过滤:拆卸装置,将三口瓶内的反应混合物趁热减压过滤,用少量热水洗涤滤渣(MnO2)。(除去二氧化锰) 3.酸化:将滤液和洗涤液合并倒入烧杯里,于冰水浴中冷却,然后在搅拌下,慢慢加入浓盐酸进行酸化(刚果红试纸检验变蓝或pH=3),至苯甲酸析出完全。 4.减压过滤:将析出的苯甲酸减压过滤,用少量冷水洗涤,挤压去水分。 【注:减压过滤前要将烧杯里的溶液进行充分的冷却。】 5.产品干燥:把制得的苯甲酸放在沸水浴上干燥。 【注:因苯甲酸在100℃左右开始升华,故应特别注意:电热套加热温度不可太高、烧杯里水量应稍多些、干燥时间长短等操作,避免干燥时局部温度过高造成苯甲酸升华而损失或熔化变成液态状。】 6.称重:产量约1.0g。 7.纯化:若要得到纯净产品,可在水中进行重结晶。 纯净的苯甲酸为白色片状或针状晶体。熔点mp=122.4℃。 七、思考题 1、在氧化反应中,影响苯甲酸产量的主要因素是哪些? 答:反应温度、甲苯与氧化剂之间的充分混合等是影响苯甲酸产量的主要因素。 2、反应完毕后,如果滤液呈紫色,为什么要加亚硫酸氢钠? 答:紫色是由过剩的高锰酸钾所致,加入亚硫酸氢钠可使高锰酸钾还原为二价的无色锰盐。
一、甲苯氯化水解法 1、工艺流程 甲苯控制条件进行侧链氯化,得到主要产物亚苄基二氯,再经酸性或碱性水 解及精馏可得苯甲醛,副产物苯甲酸。 酸性水解可用硫酸、磷酸、盐酸或甲酸等,并以锌或铁等金属盐为催化剂,如氢氧化锌、磷酸锌、月桂酸锌等,用量约为亚苄基二氯的 0. 05% ; 碱性水解 主要用碳酸钠(有的工厂用有几件替代可提高收率),在 70 ~ 80 ℃下水解 5 ~ 6 h,苯甲醛的收率为 96% ~ 97% 。 2、问题 A.水解法的废液处理有待解决 B.反应过程产生大量的氯化氢容易腐蚀设备及管道,对材质要求很高 C.产品含氯,不能直接应用于药品、香料的合成,必须增加产品精制工段,提高了产品成本 3、杭州电化集团的工艺改进 杭州电化集团有限公司所采用的新工艺是:甲苯侧链光照氯化生成二氯苄, 控制三氯苄的生成量,通过精馏分离除去一氯苄( 循环套用),水解二氯苄含量 高的馏分得到粗苯甲醛 ,经蒸馏得高纯度的苯甲醛产品(≥99.5%)。 文章(《苯甲醛生产技术剖析》邵洪根)详细给出了生产流程及流程中的重 要控制点。
二、甲苯液相氧化法 1. 钴盐为催化剂、溴化物为催化助剂、空气为氧源的液相氧化工艺
此工艺中苯甲醛作为副产物生产,经常出现在以甲苯为原料生产己内酰胺(意大利SNIA工艺)、苯甲酸的工艺流程中。国外早已工业化,国内没有使用此法将苯甲醛作为主产品的生产厂家。 优点:产品不含氯,应用范围广 缺点:氧化工艺不好控制,甲苯很容易被过度氧化成苯甲酸;产品中杂质较多,除苯甲醇、苯甲酸外还存在苯甲酸苄酯等酯类化合物。而且,甲苯的单程转化率不超过20%,若要提高苯甲醛的选择性还需要进一步降低甲苯转化率到个位数水平,增加了生产中的动力消耗 改进措施: A.可以通过加入惰性气体的方式控制氧源中氧气的浓度防止过度氧化 B.降低反应温度,减少物料在反应器中的停留时间 C.在反应体系中加入一种或多种脂肪族或芳香族的含氮化合物,提高苯甲醛在反应产物中的分布 2. 三氧化二锰法绿色氧化工艺 利用二氧化锰在 650 ℃下灼烧得到三氧化二锰,使用该原料与中等浓度的硫酸与甲苯在反应器内进行固、油、水三相反应,甲苯氧化成苯甲醛。油相蒸馏回收,分离出苯甲醛成品和没有反应的甲苯,甲苯用于循环使用;水相经活性炭吸附处理循环使用;固相副产物( 主要是硫酸锰) 可作为成品出售。用此方法制备苯甲醛的最高收率为 91%。 此法是甲苯间接电氧化法的改进版,是一种具有挑战性的方法。
羟基苯甲醛的精细合成工艺研究 通过对羟基苯甲醛的合成工艺研究,能够进一步的确定羟基苯甲醛的工艺条件。以工艺生产开展状况特点进行试验,能够对羟基苯甲醛中间产物生成以及整体工艺效果进行确认。本文对羟基苯甲醛的精细合成工艺进行研究。 标签:羟基苯甲醛;精细;合成工艺 对羟基苯甲醛的研究需要确认基本特点,能够根据化工生产的需求进一步的实现精细化合成效果的提升。对于羟基苯甲醛的研究是化工生产工艺水平提升的关键。 1羟基苯甲醛特点 羟基苯甲醛有3种异构体,即邻羟基苯甲醛、对羟基苯甲醛和间羟基苯甲醛,对羟基苯甲醛又名对甲醛苯酚。从水中析出者为白色至浅黄色针状结晶。有芳香气味。在常压下可升华而不分解。分子量122.12。熔点115~116℃。相对密度1.129 (130/4℃)。折射率1.5705(130℃)。微溶于水和苯,易溶于乙醇、乙醚、丙酮、乙酸乙酯,30.5℃时在水中的溶解度为1.38,65℃时在苯中的溶解度为3.68。小鼠腹腔注射LD50500mg/kg。对羟基苯甲醛是医药、香料、液晶的重要中间体,与硫酸二甲酯反应可制得茴香醛,与乙醛作用可制得对羟基肉桂醛,进一步氧化可制得肉桂酸,本品直接氧化可制对羟基苯甲酸,还原制对羟基苯甲醇等,均可用作香料;医药中间体;液晶原料;其他有机合成中间体,用途较广泛。间羟基苯甲醛除直接用作香料外,还用制作其他香料的中间体;医药原料,生产盐酸脱羟肾上腺素、肾上腺素、奎宁等;镀镍光亮剂;化学分析试剂(糖定量分析);照相乳剂及杀菌剂等。 邻羟基苯甲醛又称水杨醛,无色透明油状液体,有特殊气味及苦杏仁味,化学性质活泼,可发生取代、缩合、氧化、维提希(Wittig)反应等。与硫酸作用呈桔红色,与金属离子可形成有色螯合物。遇三氯化铁溶液显紫色。可被还原成水杨醇。主要用于生产香料“香豆素”及“二氢香豆素”的原料,配制紫罗兰香料,还可用作杀菌剂。 对羟基苯甲醛制备方法:由苯酚为原料,使氯仿与苯酚钠盐在60℃反应。或由苯酚与三氯乙醛在碳酸钾催化下缩合,再经甲醇钠分解。还可在三氯化铝催化剂下将干燥氯化氢通入苯酚与氢氰酸的混合液,反应后再在冰水中分解制取对羟基苯甲醛。 2实验室制备对羟基苯甲醛方法 以苯酚和三氯甲烷为原料,在碱性溶液中加热,进行Reimer-Tiemann反应,同时生成对羟基苯甲醛及少量水杨醛(邻羟基苯甲醛)。在50 mL烧瓶中加入e (2 g,8 mmo1)和a(o.94 g,8 mmo|),以及20 mL乙醇,滴加几滴哌啶,回
实验 苯甲酸乙酯的制备 一、实验目的: 1、掌握酯化反应原理,苯甲酸乙酯的制备方法,了解三元共沸除水原理。 2、复习分水器的使用及液体有机化合物的精制方法。 3、进一步练习蒸馏、萃取、干燥和折光率的测定等基本操作。 二、实验原理: 苯甲酸,乙醇在浓硫酸的催化下进行酯化反应,生成苯甲酸乙酯与水。 反应机理: 由于苯甲酸乙酯的沸点较高,很难蒸出,所以本 实验采用加入环己烷的方法,使环己烷、乙醇和水形 成三元共沸物,其沸点为℃。三元共沸物经过冷却形 成两相,使环己烷在上层的比例大,再回反应瓶,而水在下层的比例大,放出下层即可除去反应生成的水,使平衡向正方向移动。 三、实验仪器及试剂: 仪器:圆底烧瓶、回流冷凝器、分液漏斗、锥形瓶、烧杯、温度计、球形冷凝管、分水器。 试剂:苯甲酸 4g 、无水乙醇10ml 、浓硫酸 3ml 、Na 2CO 3、环己烷8ml 、乙醚、无水MgSO 4、沸石。 装置图: 反应装置 蒸馏装置 四、实验步骤: 1、加料:于50ml 圆底烧瓶中加入:4g 苯甲酸;10ml 乙醇;8ml 环己烷;3ml 浓硫酸,摇匀,加沸石。按照实验仪器左图组装好仪器(安装分水器),加热反应瓶,开始回流。 2、分水回流:开始时回流要慢,随着回流的进行,分水器中出现上下两层。当下层 试剂 d 420 ./℃ n D 20 乙醇 苯甲酸 249 环己烷 80 乙醚 苯甲酸乙酯 211~213
接近分水器支管时将下层液体放入量筒中。继续蒸馏,蒸出过量的乙醇和环己烷,至瓶内有白烟或回流下来液体无滴状(约2h ),停止加热。 3、中和:将反应液倒入盛有30mL 水的烧杯中,分批加入碳酸钠粉末至溶液呈中性(或弱碱性),无二氧化碳逸出,用PH 试纸检验。 4、分离萃取、干燥、蒸馏:用分液漏斗分出有机层,水层用25mL 乙醚萃取,然后合并至有几层。用无水MgSO 4干燥,粗产物进行蒸馏,低温蒸出乙醚。当温度超过140℃时,用牛角管直接接收210~213℃的馏分。 5、检验鉴定: 物理方法:取少量样品,用手扇动其,在闻其气味,应该稍有水果气味。 化学方法:酯与羟胺反应生成一种氧酸。氧酸与铁离子形成牢固的品红色的络合物。在试管中加入两滴新制备的酯,再加入5滴溴水。有溴水的颜色不变或没有白色沉淀生成,将5滴新制备的酯滴入干燥的试管中,在加入7滴3%的盐酸羟胺的95%酒精溶液和3滴2%的NaOH 溶液,摇匀后滴入7滴5%HCl 溶液和1滴5% FeCl3溶液,试管内显示品红色,证明酯的存在。 色谱分析:查找相关苯甲酸乙酯的色谱图,在分析产品的色谱与之对照。可以证明苯甲酸乙酯存在与否。 五、实验记录及处理: 所加试剂的量: 收集到产品的量: 参考:苯甲酸质量m 1=4g 摩尔质量 M 1=122g/mol 产物苯甲酸乙酯摩尔质量 M 2=150g/mol 实验中乙醇原料过量,苯甲酸设为完全反应,则理论苯甲酸乙酯产物量为 m 产物=4 x 150/122 g=4.918g ρ产物=1.046g/ml V 理论= m 产物÷ρ产物=÷= V 实际= 产率= V 实际÷V 理论=÷=% 误差分析:①开始分流是没调节好温度,使蒸汽流至蒸馏烧瓶下端管内。②萃取是不慎将试液流出,使产物减少。 六、思考与讨论: 1、本实验采用何种措施提高酯的产率 2、为什么采用分水器除水 3、何种原料过量为什么为什么要加苯 4、浓硫酸的作用是什么常用酯化反应的催化剂有哪些 5、为什么用水浴加热回流 6、在萃取和分液时,两相之间有时出现絮状物或乳浊液,难以分层,如何解决 七、注意事项: 1、注意浓硫酸的取用安全。加入浓硫酸应慢加且混合均匀,防止炭化。 2、回流时温度和时间的控制(反应初期小火加热、反应终点的正确判断)。 3、分水回流开始要控制温度,控制先前一个小时保持回流蒸汽在分水器接圆底烧瓶内管处。 结果与讨论 1、实验数据记录及处理 苯甲酸质量m1=8g 摩尔质量 M1=122g/mol 产物苯甲酸乙酯摩尔质量 M2=150g/mol 实得产物苯甲酸乙酯 ml 产物为无色透明液体,有芳香气味。 实验中乙醇原料过量,苯甲酸设为完全反应,则理论苯甲酸乙酯产物量为 m 产物=8 x 150/122 g= ρ产物=ml V 理论= m 产物÷ρ产物=÷= V 实际= 产率= V 实际÷V 理论=÷=% 结果分析:本次实验产率算比较高。 2、造成产率降低的原因分析: ①开始分流时温度没调节好,温度
苯甲酸和苯甲醛的合成暨Cannizzaro反应作者:段璞 摘要:本文是第四次合成化学实验的实验报告,主要介绍了以苯甲醛为原料进行的Cannizzaro反应制备苯甲酸与苯甲醇的方法。反应后通过萃取分离苯甲酸与苯甲醇,并分别运用重结晶和加压蒸馏提纯两产物。本次实验投放原料苯甲醛10mL(10.4g,0.1mol),共制得提纯产物苯甲酸4.38g、苯甲醇3.68g。 实验原理: 无-α-氢的醛(如芳香醛、甲醛等)在浓碱的作用下发生歧化反应,一分子醛失去氢被氧化为酸,而另一分子醛得到氢而被还原为醇,此反应即为Cannizzaro反应。 如本次反应中使用的苯甲醛歧化反应: Cannizzaro反应的实质是羰基的亲核加成。反应的机理是OH-对一个醛分子的羰基进行亲核进攻生成负离子,此负离子使原来醛基上的氢带着一对电子对另一分子芳香醛进行亲核加成。反应机理如下:
由于第一步反应为可逆反应,且平衡偏向反应物一侧,所以Cannizzaro反应需要非常高的碱度,常用过量一倍以上的浓度为50%的强碱。本次反应中使用了10g氢氧化钠(0.25mol,即2.5当量)。 利用产物苯甲酸与苯甲醇在碱性溶液与乙醚中溶解度的不同(苯甲酸以苯甲酸盐的形式存在于碱液,而苯甲醇主要存在于乙醚)达到分离两种产物的效果。接着利用未反应完全的苯甲醛易与亚硫酸氢钠反应生成α-羟基苄磺酸钠,从而达到与苯甲醇分离的效果。反应机理如下: 实验操作及现象: 1.于150mL锥形瓶中称取10g氢氧化钠(0.25mol)加入10mL水。溶解并冷却至室温后, 边振摇边加入10mL苯甲醛(10.4g,0.1mol)。加完后用称量纸封口,剧烈振摇,充分混合,数分钟后,原料凝结成固体,呈现微黄色,后逐渐变为淡粉色。于避光处放置两礼拜。 2.取适量水溶解,转移至分液漏斗,乙醚萃取3次,每次25mL。初次醚层呈红褐色,水 层微浑浊,萃取若干次后基本澄清。合并醚层,倒入分液漏斗中,先用10mL饱和亚硫
苯甲醇和苯甲酸的制备实验 一、实验原理 利用坎尼扎罗(Cannizzaro)反应由苯甲醛制备苯甲醇和苯甲酸。 坎尼扎罗反应是指无α-活泼氢的醛类在浓的强NaOH或KOH水或醇溶液作用下发生的歧化反应。此反应的特征是醛自身同时发生氧化及还原作用,一分子醛被氧化成羧酸(在碱性溶液中成为羧酸酸盐),另一分子醛则被还原成醇。 主反应: 机理:醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢 负离子的形式转移到另一分子的羰基不能碳原子上。 二、反应试剂、产物、副产物的物理常数
三、药品 四、实验流程图 五、实验装置图
图1 磁力搅拌器图2 分液漏斗的振摇方法图3 分液漏斗图4 抽滤装置 六、实验内容 往锥形瓶中加 12.0g(0.21mol)氢氧化钾和12ml水,放在磁力搅拌器上搅拌,使氢氧化钾溶解并冷至室温。在搅拌的同时分批加入新蒸过的苯甲醛,每次加入2-3ml,共加入13.5ml(约14g,0.13mol)。加后应塞紧瓶口,若锥形瓶内温度过高,需适时冷却。继续搅拌60min,最后反应混合物变成白色蜡糊状。 (1)苯甲醇 向反应瓶中加入大约45ml水,使反应混合物中的苯甲酸盐溶解,转移至分液漏斗中,用45ml 乙醚分三次萃取苯甲醇,合并乙醚萃取液。保存水溶液留用。 依次用15ml25%亚硫酸氢钠溶液及8ml水洗涤乙醚溶液,用无水硫酸镁干燥。水浴蒸去乙 醚后,继续蒸馏,收集产品,沸程204-206℃,产率为75%。 纯苯甲醇有苦杏仁味的无色透明液体。沸点bp=205.4℃,折光率=1.5463。 (2)苯甲酸 在不断搅拌下,往留下的水溶液中加入浓盐酸酸化,加入的酸量以能使刚果红试纸由红变 蓝为宜。充分冷却抽滤,得粗产物。 粗产物用水重结晶后晾干,产率可达80%。 纯苯甲酸为白色片状或针状晶体。熔点mp=122.4℃。 (一)制备阶段 1.准备锥形瓶:一只100ml锥形瓶。 2.加药品与歧化反应:向锥形瓶中加 12.0g氢氧化钾和12ml水,向瓶内放入一只搅拌子, 然后将锥形瓶放在磁力搅拌器上搅拌,使氢氧化钾溶解并冷至室温。在搅拌的同时分批加入新蒸过的苯甲醛,每次加入2-3ml,共加入13.5ml(约14g,0.13mol)。加后应塞紧瓶口,若锥形瓶内温度过高,需适时冷却。继续搅拌60min,最后反应混合物变成白色蜡糊状。 【为避免歧化反应过快产生大量热量,造成温度过高增加氧化副反应,故需将苯甲醛分几批加入】 (二)后处理阶段 1.分离苯甲醇 (1)加水溶解:向反应瓶中加入大约45ml水,使反应混合物中的苯甲酸盐溶解,转移至 分液漏斗中。 【在磁力搅拌器上尽量搅拌时间长一些,以保障苯甲酸钾盐充分溶解在水中,减少与苯甲 醇分子的包裹,有利于下一步的乙醚萃取】