酵母单杂分析
酵母单杂交技术是体外分析DNA与细胞内蛋白质相互作用的一种方法,通过对酵母细胞内报告基因表达状况的分析来鉴别DNA结合位点并发现潜在的结合蛋白基因,或对结合位点进行分析。运用此技术能筛选到与DNA结合的蛋白质,并可直接从基因文库中得到编码该蛋白质的核苷酸序列而无需复杂的蛋白质分离纯化操作,故在蛋白质研究中具有一定的优势;而且酵母属真核细胞,通过酵母系统得到的结果比其它体外技术获得的结果更能体现真核细胞内基因表达调控的情况。
【实验目的】
了解酵母单杂交的基本原理和应用,掌握酵母单杂交的主要步骤及注意事项,学会酵母感受态的制作与转化,基因文库的构建及筛选。
【实验原理】
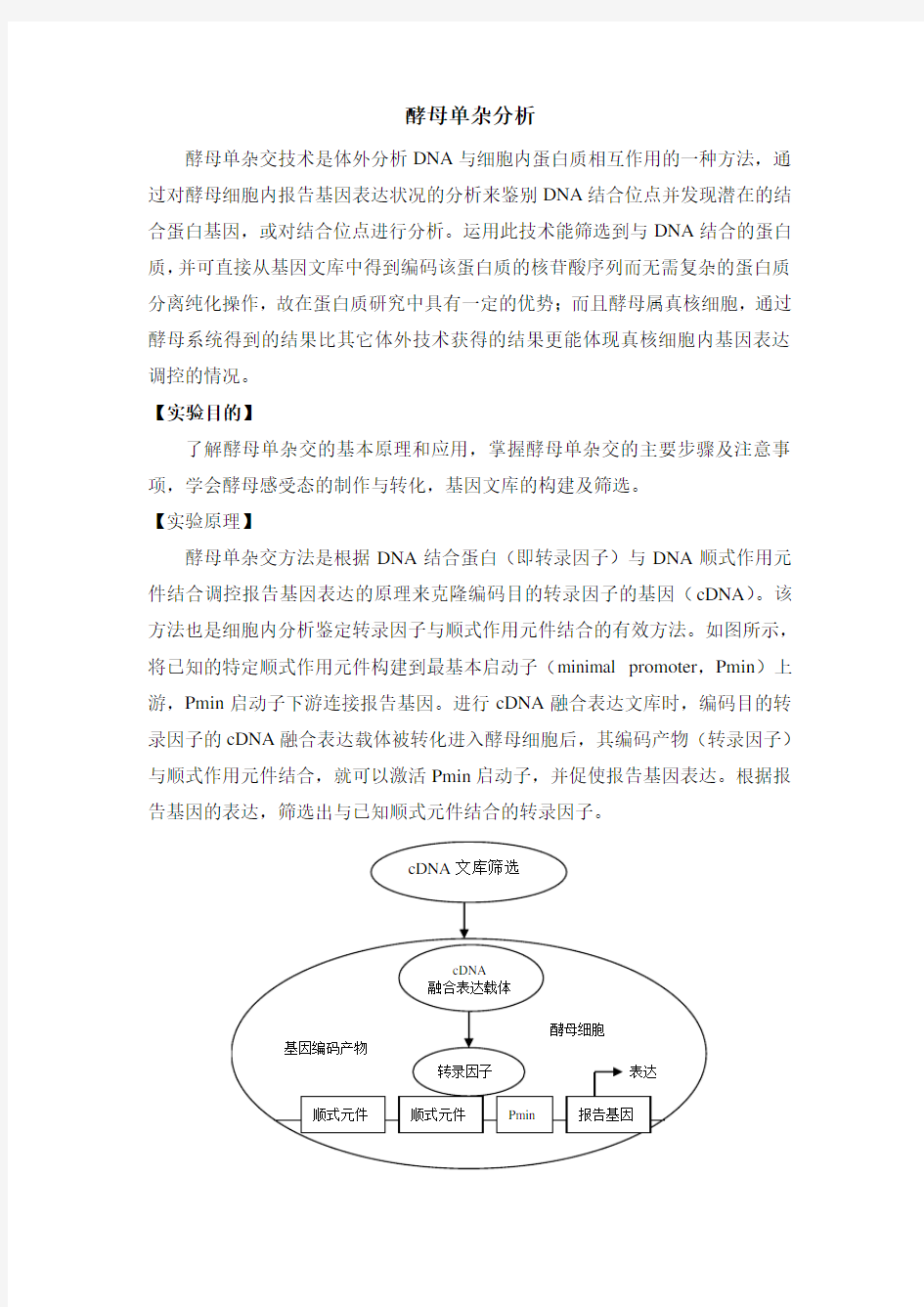
酵母单杂交方法是根据DNA结合蛋白(即转录因子)与DNA顺式作用元件结合调控报告基因表达的原理来克隆编码目的转录因子的基因(cDNA)。该方法也是细胞内分析鉴定转录因子与顺式作用元件结合的有效方法。如图所示,将已知的特定顺式作用元件构建到最基本启动子(minimal promoter,Pmin)上游,Pmin启动子下游连接报告基因。进行cDNA融合表达文库时,编码目的转录因子的cDNA融合表达载体被转化进入酵母细胞后,其编码产物(转录因子)与顺式作用元件结合,就可以激活Pmin启动子,并促使报告基因表达。根据报告基因的表达,筛选出与已知顺式元件结合的转录因子。
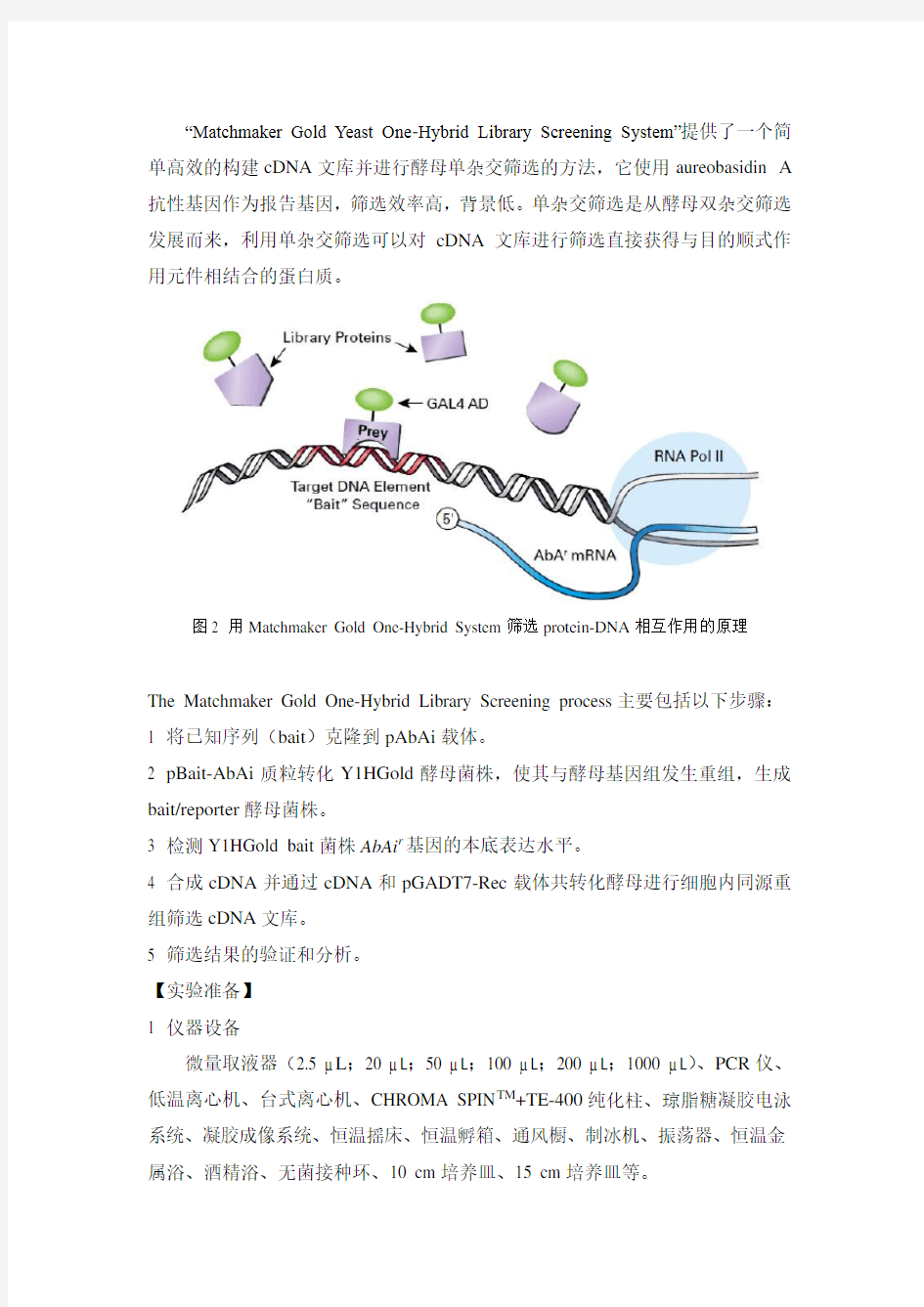
“Matchmaker Gold Yeast One-Hybrid Library Screening System”提供了一个简单高效的构建cDNA文库并进行酵母单杂交筛选的方法,它使用aureobasidin A 抗性基因作为报告基因,筛选效率高,背景低。单杂交筛选是从酵母双杂交筛选发展而来,利用单杂交筛选可以对cDNA文库进行筛选直接获得与目的顺式作用元件相结合的蛋白质。
图2 用Matchmaker Gold One-Hybrid System筛选protein-DNA相互作用的原理
The Matchmaker Gold One-Hybrid Library Screening process主要包括以下步骤:
1 将已知序列(bait)克隆到pAbAi载体。
2 pBait-AbAi质粒转化Y1HGold酵母菌株,使其与酵母基因组发生重组,生成bait/reporter酵母菌株。
3 检测Y1HGold bait菌株AbAi r基因的本底表达水平。
4 合成cDNA并通过cDNA和pGADT7-Rec载体共转化酵母进行细胞内同源重组筛选cDNA文库。
5 筛选结果的验证和分析。
【实验准备】
1 仪器设备
微量取液器(2.5 μL;20 μL;50 μL;100 μL;200 μL;1000 μL)、PCR仪、低温离心机、台式离心机、CHROMA SPIN TM+TE-400纯化柱、琼脂糖凝胶电泳系统、凝胶成像系统、恒温摇床、恒温孵箱、通风橱、制冰机、振荡器、恒温金属浴、酒精浴、无菌接种环、10 cm培养皿、15 cm培养皿等。
2 实验材料
Y1HGold酵母株、TOP10大肠杆菌菌株、各种方法提取的RNA、pGADT7-Rec 质粒和pBait-AbAi质粒等。
3 主要试剂
(1)Advantage 2 PCR试剂盒、Easy Yeast Plasmid Isolation试剂盒、Matchmaker Insert Check PCR Mix 1、Matchmaker Insert Check PCR Mix 2。
(2)各种基础培养基和营养缺陷培养基:minimal SD base,minimal SD agar base,YPD medium,YPD agar medium,-Leu DO supplement,-Ura DO supplement,腺苷酸(adenine),PEG8000,carring DNA,aureobasidin A,LB培养基,氨苄西林。
(3)NaCl溶液(0.9%)、sodium acetate (3 mol/L)、50% PEG、10×LiAc(1 mol/L)、10×TE buffer、DTT(100 mmol/L)、RNaseH、限制性内切酶。【实验方法】
1 pBait-AbAi载体的构建(酵母报道子的构建)
注:酵母报道子(pBait-AbAi)包含目的顺式作用元件的一个或多个拷贝,且插入到pAbAi载体AbAi r报告基因的上游。大量研究表明最有效的构建应包含目的DNA三个以上的首尾连接的拷贝。首尾连接的拷贝产生方式很多,但对于长度小于20 bp的调控元件,人工合成寡核苷酸是最方便可靠的途径。
(1)设计并合成包含目的序列的两条反向平行的寡核苷酸序列,且两端加上与pAbAi载体酶切产物一致的粘性末端(建议合成一个目的序列的突变序列作为对照,以排除可能的假阳性)。
(2)用TE buffer溶解寡核苷酸至终浓度100 μmol/L。
(3)将正向链和反向链按照1:1的比例混合(退火后的双链寡核苷酸最大浓度为50 μmol/L)。
(4)95 ?C保温30 s,去除二级结构。
(5)72 ?C保温2 min,37 ?C保温2 min,25 ?C保温2min。
注:缓慢退火,有助于双链寡核苷酸的形成。
(6)冰上放置。退火后的产物可贮存在-20 ?C冰箱备用。
(7)酶切1 μL pAbAi载体,用凝胶回收纯化或柱纯化的方式纯化酶切产物。
注:回收前,可用琼脂糖凝胶检测是否酶切完全。
(8)将退火后的寡核苷酸稀释100倍至终浓度为0.5 μmol/L。
(9)在连接反应管中加入如下成分:
pAbAi载体(50 ng/μL) 1.0 μL
annealed oligonucleotide (0.5 μmol/L) 1.0 μL
10×T4 DNA ligase buffer 1.5 μL
BSA(10 mg/mL)0.5 μL
Nuclease-free H2O 10.5 μL
T4 DNA ligase (400 U/μL)0.5 μL
总体积15 μL
注:如果有必要,可用1 μL nuclease-free H2O代替寡核苷酸作为阴性对照。
(10)将反应体系室温放置连接3 h,转化E coli,采用常规方法检测阳性克隆。
注:可用酶切或测序进行检测。
2 质粒转化酵母细胞,生成Bait-Reporter酵母菌株(图)
图3 Bait-Reporter酵母菌株生成原理示意图
(1)用BstB I或者Bbs I酶切2 μL pBait-AbAi,pMutant-AbAi,p53-AbAi质粒,使其在URA3基因处断开,纯化酶切产物。
(2)按Matchmaker Yeast Transformation System 2的步骤用1 μl酶切后的质粒转化Y1HGold酵母。
(3)稀释每个转化体系至1/10、1/100、1/1000,分别取每个稀释物均匀涂于SD/-Ura琼脂平板上。3 d后挑取5个单克隆,用Matchmaker Insert Check PCR Mix1进行PCR检测阳性克隆,用Y1HGold的单克隆做阴性对照。(4)在PCR管中加25 μl PCR-grade H2O。
(5)用干净的枪头轻轻接触酵母单克隆,以获得非常少量的酵母细胞。将枪头伸进PCR-grade H2O中搅拌,使酵母细胞散开。
注:切忌挑取整个酵母单克隆,因为细胞过多会阻止PCR反应的进行。
如果加入细胞后水变浑浊,证明加入了过多的酵母细胞。
(6)向每个管中加入25 μl Matchmaker Insert Check PCR Mix,混匀,离心。每个PCR管中现已含有如下反应物:
Matchmaker Insert Check PCR Mix 25 μl
H2O/yeast 25 μl
总体积50 μl
(7)按下述程序进行PCR反应;
95 ?C 1 min
98 ?C 10 s
55 ?C 30 s 30 cycles
68 ?C 2 min
(8)取5 μl PCR产物,用1%的琼脂糖凝胶电泳分析。
注:引物与AbA基因以及URA3下游的Y1HGold基因组结合,扩增片段长约1.4 kb。
图4 PCR检测pBait-AbAi的插入情况
正确的PCR检测结果应是:
阳性对照:1.4 kb
阴性对照:无条带
诱饵菌株:1.35 kb+insert size
(9)分别挑取PCR检测呈阳性的诱饵克隆和p53-AbAi对照克隆,在SD/-Ura 平板上划线培养。30 ?C孵育3 d后,将平板置于4 ?C保存,即为新构建的Y1HGold[Bait/AbAi]菌株和[p53/AbAi]对照菌株。
(10)经过长期放置后,挑取单克隆在YPDA液体培养基中过夜培养,离心收集菌体,用 1 mL预冷培养基(100 ml灭菌的YPDA与50 ml灭菌的75%甘油混合)重悬菌体,速冻后与-70 ?C保存。
3 检测诱饵菌株AbA r基因的表达
在不存在捕获物的情况下,由于克隆到pAbAi载体中的诱饵序列不同,诱饵菌株报告基因的本底表达水平也不相同。例如:p53-AbAi对照的最低aureobasidin A抑制浓度为100 ng/ml。
注:酵母单杂交实验成功的前提是没有内源转录因子能够与目的序列结合或者结合能力非常弱。因此在进行文库筛选之前,检测所构建的诱饵菌株AbA r基因的表达情况十分重要。所以需要进行实验以确定进行文库筛选时抑制诱饵菌株报告基因本底表达所需的AbA浓度。
(1)分别挑取诱饵克隆和对照克隆,用0.9% NaCl重悬细胞,调节A600到0.002(大约2000个细胞/100 μl)。
(2)在下述培养基上分别涂布100 μl重悬后的菌液,30 ?C培养2-3 d。
SD/-Ura
SD/-Ura with AbA (100 ng/mL)
SD/-Ura with AbA (150 ng/mL)
SD/-Ura with AbA (200 ng/mL)
预期结果如下所示:
表1 AbA r基因预期本底表达结果
[AbA]/(ng/mL)Y1HGold[p53-AbAi]克隆数Y1HGold[pBait-AbAi]克隆数
0 约2000 约2000
100 0 Bait dependent
150 0 Bait dependent
200 0 Bait dependent
(3)在进行文库筛选时,使用AbA的浓度应为最低抑制浓度,或使用比最低抑制浓度稍高的AbA浓度(高约50-100 ng/mL),以彻底抑制诱饵菌株的生长。
注:如果200 ng/mL AbA不能抑制本底表达,可以尝试提高AbA浓度至500-1000 ng/mL。然而,在不存在捕获物的情况下,如果1000 ng/mL的AbA浓度仍无法抑制AbA r基因的表达,那么很可能存在能够识别并与目的序列结合的内源调控因子,因而该目的序列无法用来进行酵母单杂交筛选。
4 文库cDNA的合成
提取试材总RNA,进行反转录合成cDNA。合成的cDNA末端具有与pGADT7-Rec相同的酶切位点。
(1)cDNA第一链的合成
①准备高质量的poly A和/或总RNA,用human placenta poly A+RNA作为阳性对照。
注:RNA的质量决定文库的质量,RNA应为所要研究的特定时期和特定组织的RNA。
②在微量离心管中加入如下反应物:
RNA模板(0.025-1.0 μg poly A和/或0.10-2.0 μg总RNA)1-2 μl CDS III(oligo-dT)or CDS III/6(random)引物 1.0 μl Deionized H2O (使总体积达到4.0 μl)1-2 μl
总体积 4.0 μl
在另外一支管中加入对照cDNA反应物,即RNA使用1 μl(1 μg)control poly A+RNA。
③72 ?C孵育2 min。
④冰上放置2 min,轻轻混匀,立即加入步骤⑤中的试剂。
⑤每一个反应加入如下试剂,轻轻混匀离心。
5×first-strand buffer 2.0 μl
DTT(100 mmol/L) 1.0 μl
dNTP mixture(10 mmol/L) 1.0 μl
SMART M-MLV RT 1.0 μl
总体积 5.0 μl
注:步骤⑤中的试剂可在步骤②之前加好置于冰上。此步是cDNA合成的起始关键步骤,变性后的RNA/引物mix冰上放置的时间不应超过2 min。
⑥如果用的是CDS III/6随机引物,25-30 ?C保温10 min。如果用的是CDS III 引物,省略此步,进行步骤⑦。
⑦42 ?C保温10 min。
⑧加入1 μl SMART III oligo,充分混合,42 ?C保温1 h。
⑨75 ?C保温10 min终止第一链的合成。
⑩降至室温,加入1 μl RNase H(2 U)。
11 37 ?C保温20 min。
12 cDNA第一链合成产物应于-20 ?C保存,可用3个月。
(2)long distance PCR (LD-PCR)合成cDNA 第二链
根据合成cDNA第一链时使用的RNA量,下表给出了进行LD-PCR时最佳的热循环数。使用的热循环数越少,非特异性PCR产物越少。
表2 RNA量与最佳热循环数
总RNA/μg Poly A+RNA/μg 循环数
1.0-
2.0 0.5-1.0 15-20
0.5-1.0 0.25-0.5 20-22
0.25-0.5 0.125-0.25 22-24
0.05-0.25 0.025-0.125 24-26
⑴LD-PCR反应混合物中加入如下物质(每个样品做2个100 μl体系,对照做1个100 μl体系):
First-strand cDNA (from protocol A) 2 μl
Deionized H2O 70 μl
10×advantage 2 PCR buffer 10 μl
50×dNTP mix 2 μl
5’RACE引物 2 μl
3’RACE引物 2 μl
Melting solution 10 μl
50×advantage 2 polymerase mix 2 μl
总体积100 μl
⑵按照以下程序进行PCR反应:
1个循环95 ?C 30 s
X个循环95 ?C 10 s
68 ?C 6 min
1个循环68 ?C 5 min
⑶取7 μl PCR产物用1.2%的琼脂糖凝胶检测,检测时使用1 kb DNA ladder。
(2)使用CHROMA SPIN+TE-400柱纯化ds cDNA
⑴为每一个要纯化的cDNA样品准备一个CHROMA SPIN+TE-400柱。
⑵将纯化柱翻转几次,充分悬浮gel matrix。
⑶移去柱的顶盖和底盖,将柱放入2 ml收集管中。
⑷将柱放入离心机,700 g离心5 min以消除平衡缓冲液,弃掉收集管中的液体。
⑸将柱放入新的收集管中,把cDNA加到gel matrix的中央,切勿使样品沿柱的内壁流下。
注:加到边上易使样品沿柱内壁流下,易混有小片段cDNA。
⑹700 g离心5 min,纯化的cDNA收集到管中。
⑺将两个纯化的cDNA样品合并到一管,测量体积。
⑻加入1/10体积3 mol/L醋酸钠(pH5.3),混匀。
⑼加入2.5倍体积无水乙醇。
⑽-20 ?C冰冻1 h。
⑾室温,14000 r/min离心20 min。
⑿小心弃去上清液,切勿碰到沉淀。
⒀14000 r/min瞬时离心,去除残留上清液。
⒁沉淀于空气中干燥10 min。
注:一般干燥至无乙醇味,不可过度干燥,否则很难溶解。
⒂用20 μl灭菌去离子水溶解沉淀,此cDNA可用来进行同源重组构建文库。纯化后的cDNA用1%的琼脂糖电泳检测。
5 构建并筛选酵母单杂交文库(cDNA融合表达文库的构建及筛选)
(1)构建并在SD/-Leu/AbA培养基上检测Y1HGold[Bait/AbAi]菌株。
注:AbA的浓度根据构建诱饵载体时转入酵母抑制本底表达时的浓度而定。(2)按SMART technology步骤合成ds cDNA,其浓度为2-5 μg/20 μL。
(3)用Yeast Transformation System 2的方法转化酵母,在转化体系中加入如下物质:
①cDNA文库转化Y1HGold[Bait/AbAi]菌株
20 μl SMART-amplified ds cDNA (2-5 μg)
6 μl pGADT7-Rec,(Sma I-linearized)(3 μg)
②Y1HGold [53/AbAi]的转化
5 μl p53 fragment (125 μg)
2 μl pGADT7-Rec,(SmaI-linearized)(1 μg)
将转化体系分别稀释至1/10、1/100、1/1000、1/10000后,各取100 μl涂100 mm平板。
文库转化涂SD/-Leu和SD/-Leu/AbA平板,对照p53转化涂SD/-Leu和SD/-Leu/AbA200平板。
(4)将剩余的所有文库转化混合物(约15 ml)涂在150 mm SD/-Leu/AbA平板上,每板涂150 μl。
(5)倒置培养3-5 d。
(6)3-5 d后,通过统计SD/-Leu 100 mm平板上的克隆数目来计算筛选的克隆
数。
注:所筛选的克隆数至少应该达到1百万,否则会降低筛选到目的产物的可能性。
筛选的克隆数=[cfu/ml on SD/-Leu]×[dilution factor]×[resuspension volume(15ml)]
例如:resuspension volume=15 ml
Plating volume=100 μl
250 colonies grew on the 1/100 dilution on SD/-Leu plates
The number of clones screened=250 cfu/0.1 m l×100×15 ml=3.75 million
(7)预期结果
阳性对照试验:SD/-Leu和SD/-Leu/AbA200培养基上的克隆数相近。
文库筛选试验:根据SD/-Leu平板上的克隆数计算所筛选的克隆数,其结果应大于1百万且SD/-Leu/AbA平板上的克隆数远远少于1百万,阳性克隆数目取决于诱饵序列。
6 阳性克隆的鉴定及cDNA质粒的分离
(1)阳性克隆重新划线培养,进行表型确认
①将阳性克隆在SD/-Leu/AbA培养基上重新划线,产生新的单克隆。
②2-4 d后,选择能够正常生长的克隆进行后续分析。
(2)酵母克隆PCR消除重复克隆
①用Matchmaker Insert Check PCR Mix 2 (Cat.No.630497)进行PCR,对插入到pGADT7载体中的cDNA片段进行扩增。PCR管中加入以下反应物:Matchmaker Insert Check PCR Mix 25 μl
H2O/Yeast 25 μl
总体积50 μl
②按下述程序进行PCR反应:
94 ?C 1 min
98 ?C 10 s
68 ?C 3 min
③PCR产物在1%的琼脂糖凝胶上进行电泳分析。产物不是单一的条带很正常,这表明在同一酵母细胞不存在一种捕获载体
注:为了确认大小相近的条带是否是同一种插入片段,用Alu I或Hae III或者其它常用的限制性内切酶消化PCR产物,产物用2%的琼脂糖凝胶进行电泳分析。
④如果大量的克隆含有同一插入片段,则另取50个克隆进行PCR分析。
⑤为了快速验证克隆,PCR产物可经过纯化后用T7引物测序。
(3)阳性cDNA质粒的分离获取
①酵母中文库质粒的分开。
与转化的大肠杆菌不同,转化的酵母细胞可以含多种相关质粒,这就意味着阳性克隆里不只含有能激活AbA r报告基因的质粒,还可能含有一种或多种不表达相互作用蛋白的cDNA质粒。如果不事先将非互作质粒分开出去而直接通过转化大肠杆菌获取质粒,那么很有可能获取到非相互作用的质粒。为了增加获取阳性克隆捕获质粒的几率,可以将阳性克隆在SD/-Leu/AbA培养基上重复涂布2-3次,每次都挑取单一的克隆进行下一步涂布。
②从酵母中获取阳性cDNA质粒
为了鉴定阳性互作相关的基因,用Easy Yeast Plasmid Isolation Kit (Cat.No.630467)从酵母中获取阳性质粒。
③转化E coli并分离阳性cDNA质粒
用常用的克隆菌株对阳性cDNA质粒进行克隆,用LB加100 g/ml ampicillin 进行选择。
(4)鉴别阳性和假阳性互作
酵母单杂交筛选可能会检测到假阳性,用以下标准可以区分阳性和假阳性阳性:正确的诱饵序列和捕获物都是激活AbA r报告基因所必需的。
假阳性:在诱饵序列突变的情况下,诱饵仍可以激活AbA r报告基因。
用下述程序在选择培养基上对阳性和假阳性相互作用进行确认:
①用Yeastmaker Transformation System 2的试剂和small-scale转化程序将100 ng 获取的捕获质粒转化到Y1HGold[Bait/AbAi]和Y1HGold[Mutant/AbAi]菌株中。注:阳性对照和阴性对照实验应该一起进行。
②在SD/-Leu和SD/-Leu/AbA培养基上涂100 μl转化混合物1/10和1/100的稀释物。
③30 ?C恒温培养3-5 d后,预期结果如表所示。
表3 阳性和假阳性相互作用验证结果
A 阳性
样品选择培养基 2 mm清晰的克隆
酵母菌SD/-Leu 有
Y1HGold[诱饵/AbAi]+靶SD/-Leu/AbA 有
酵母菌SD/-Leu 有
Y1HGold[突变/AbAi]+靶SD/-Leu/AbA 无(或者很小)
B 假阳性
样品选择培养基 2 mm清晰的克隆
酵母菌SD/-Leu 有
Y1HGold[诱饵/AbAi]+靶SD/-Leu/AbA 有
酵母菌SD/-Leu 有
Y1HGold[突变/AbAi]+靶SD/-Leu/AbA 有
(5)阳性克隆的测序分析
一旦相互作用被验证为阳性,就可以测序鉴定捕获载体的插入cDNA片段,验证与GAL4 AD序列融合的开放阅读框(ORF)序列,并与GenBank、EMBL 或其他数据库中的序列进行比较。
【注意事项与建议】
1 进行一轮酵母单杂交筛选后,得到的阳性克隆可能非常少或者非常多,在这种情况下,建议做如下处理。
⑴阳性克隆太少:
检查所筛选的克隆数是否大于1 million
通过阳性对照和阴性对照检查培养基是否正常
重新检测诱饵的最低AbA抑制浓度
试着增加目的序列的拷贝数,通常目的序列的拷贝数为3时,试验效果最好。
⑵阳性克隆太多:
检查是否使用了最佳AbA抑制浓度;如果使用了100 ng/ml AbA,使用200 ng/ml 的AbA浓度重新筛选
通过阳性对照和阴性对照检查培养基营养缺陷是否正常
可能文库中存在大量能编码与诱饵序列结合蛋白的cDNA。可通过酵母PCR将其克隆分类,每一类中的代表可用来进行阳性互作分析。
2 对阳性克隆进行测序之前需进行以下试验。
⑴用新鲜的选择性培养基对阳性克隆重新划线培养,进行表型确认;
⑵酵母克隆PCR,对重复的克隆进行分类;
⑶阳性cDNA质粒的分离;
⑷阳性和假阳性互作的辨别。
酵母双杂交系统 1.原理 酵母双杂交系统的建立得力于对真核细胞调控转录起始过程的认识。研究发现,许多真核生物的转录激活因子都是由两个可以分开的、功能上相互独立的结构域(domain)组成的。例如,酵母的转录激活因子GAL4,在N端有一个由147个氨基酸组成的DNA结合域(DNA binding domain,BD),C端有一个由113个氨基酸组成的转录激活域(transcription activation domain,AD)。GAL4分子的DNA结合域可以和上游激活序列(upstream activating sequence,UAS)结合,而转录激活域则能激活UAS下游的基因进行转录。但是,单独的DNA结合域不能激活基因转录,单独的转录激活域也不能激活UAS 的下游基因,它们之间只有通过某种方式结合在一起才具有完整的转录激活因子的功能。 2.试验流程 酵母双杂交系统正是利用了GAL4的功能特点,通过两个杂交蛋白在酵母细胞中的相互结合及对报告基因的转录激活来捕获新的蛋白质,其大致步骤为: 2.1、视已知蛋白的cDNA序列为诱饵(bait),将其与DNA结合域融合,构建成诱饵质粒。 2.2、将待筛选蛋白的cDNA序列与转录激活域融合,构建成文库质粒。2.3、将这两个质粒共转化于酵母细胞中。 2.4、酵母细胞中,已分离的DNA结合域和转录激活域不会相互作用,但诱饵蛋白若能与待筛选的未知蛋白特异性地相互作用,则可激活报告基因的转录;反之,则不能。利用4种报告基因的表达,便可捕捉到新的蛋白质。 3.特点 优点 蛋白--蛋白相互作用是细胞进行一切代谢活动的基础。酵母双杂交系统的建立为研究这一问题提供了有利的手段和方法。 缺点
2.1.1酵母双杂交 2.1.1.1Gateway入门克隆 设计Gateway引物时,在上游引物的5'端加上B1序列:GGGG-ACA-AGT-TT G-TAC -AAA-AAA-GCA-GGC-TNN-,下游引物的5'端加上B2序列:GGGG-ACC-ACT-TT G-T AC-AAG-AAA-GCT-GGG-TN-。其中,5'-GGGG序列是保护碱基,防止引物的重要部分被降解,下划线加粗的部分是在整个的Gateway克隆中可以保存下来的序列,3'端的碱基N是为了保证经过入门载体构建目的载体时阅读框的正确性,一般建议为C。 通过PCR扩增获得带有att B位点的基因片段,扩增体系和条件见3.2.2.2,其中将退火温度改为65℃。获得扩增产物后对其进行回收纯化,测定纯化后DNA 的质量和浓度后进行下一步的BP反应,反应体系如下: att B-PCR产物(≥10 ng/μL)1-7μL pDONR221 (150 ng/μL)1μL TE buffer, pH 8.0 补足8μL 将上述混合物加入离心管中,加入2μL BP反应酶,加入之前需将其在涡旋仪上轻轻振荡两次,所有组分混匀离心后,25℃反应1h,加入1μL蛋白酶K后,混匀离心,37℃反应10min终止BP反应,将BP反应产物参照3.2.2.6进行转化,由于pDONR221载体为Kan抗性,所以选用含有50μg/mL Kan抗生素的LB平板进行阳性克隆筛选,参照3.2.2.7检测阳性克隆,然后根据3.2.2.8中的方法提取重组质粒,测定质量和浓度后送至测序公司进行测序。 进行BP反应时,需注意以下几项: (1)对于BP反应来说,最高效的是采用线性的att B-PCR产物和超螺旋的att P 入门载体; (2)为了提高BP反应的效率,可以将建议的25℃反应1h适当延长至4-6h,可 以将效率提高2-3倍,或者延长至过夜反应,可以将效率提高5-10倍,对 于长片段克隆来讲,适当的延长反应时间是非常必要的; (3)提高体系中PCR产物的量可以增加反应效率,但每10μL体系中PCR产物 最好不要超过250ng。 2.1.1.2诱饵载体构建 重组的入门载体测序正确后,通过LR反应来构建酵母双杂交的诱饵载体,LR反应体系如下: 入门载体(50-150 ng)1-7μL pDEST32 (150 ng/μL)1μL TE buffer, pH 8.0 补足8μL
酵母单杂交 —研究蛋白质与特定DNA序列之间的相互作用 酵母单杂交技术最早是从酵母双杂交技术发展而来的,酵母双杂交技术通过对报告基因的表型进行检测以实现对蛋白质间相互作用的研究,而酵母单杂交技术则通过对报告基因的表型检测,分析DNA与蛋白之间的相互作用,以研究真核细胞内的基因表达调控。 ●顺式作用元件 存在于基因旁侧序列中能影响基因表达的序列,本身不编码任何蛋白质,仅仅提供一个作用位点,与反式作用因子相互作用参与基因表达调控。 ●反式作用因子 是指能直接或间接地识别或结合在顺式作用元件核心序列上参与调控靶基因转录效率的蛋白质。 酵母单杂交(Yeast one-hybrid)是根据DNA结合蛋白(即转录因子)与DNA顺式作用元件结合调控报道基因表达的原理,克隆与靶元件特异结合的转录因子基因(cDNA)的有效方法。 其理论基础是:许多真核生物的转录激活子由物理和功能上独立的DNA结合区(DNA-binding domain BD)和转录激活区(Activationdomain AD)组成。BD可识别DNA上的特异序列,并使转录激活结构域定位于所调节的基因的上游;AD可同转录复合体的其他成分作用,启动它所调节的基因的转录。两个结构域各据功能,互不影响。 (单独的BD虽然能和启动子结合,但是不能激活转录。) 这两个结构域各具功能,互不影响,单独存在时没有转录激活的功能,只有两者通过共价或非共价键连接建立起来的空间结构方可表现出一个完整的激活特定基因表达的激活因子的功能。 用于酵母单杂交系统的酵母GAL4蛋白即是一种典型的转录因子。研究表明GAL4 的DNA结合结构域靠近羧基端,含有几个锌指结构,可激活酵母半乳糖苷酶的上游激活位点(UAS);而转录激活结构域可与RNA 聚合酶或转录因子TFIID相互作用,提高RNA 聚合酶的活性。在这一过程中,DNA 结合结构域和转录激活结构域可完全独立地发挥作用。 据此,我们可将GAL4 的DNA结合结构域置换为其他蛋白,只要他能与我们想要了解的目的基因相互作用,就照样可以通过其转录激活结构域激活RNA聚合酶,从而启动对下游报告基因的转录。
目录 (一)介绍 4 (二)试剂盒物品清单 7 (三)额外附加物品列表9 (四)酵母菌株11 (五)酵母载体14 (六)方法简述:单杂交文库的构建和筛选16 方法简述:双杂交文库的构建和筛选17 (七)构建用于酵母单杂交的报告质粒载体18 (八)构建用于酵母双杂交的DNA-BD融合载体19 (九)构建生成cDNA文库21 (十)构建和筛选酵母单杂交和双杂交文库(简述)27 (十一)酵母单杂交文库的构建和筛选28 (十二)酵母双杂交文库的构建和筛选30 方法A:通过酵母配对(Yeast Mating)来筛选目的蛋白30 方法B:通过共转化的方法筛选目的蛋白35 (十三)分析阳性相互作用结果38 (十四)问题解决指南44 (十五)参考文献47 (十六)相关产品50 附录A: 双链 cDNA合成的典型结果51 附录B: 酵母感受态的制备—LiAc 法52 附录C:单杂交对照载体信息53 附录D:双杂对照载体信息54 表格列表 Table I. BD Matchmaker酵母菌株的基因型11 Table II. BD Matchmaker酵母菌株的表型11 Table III.单杂交系统的载体14 Table IV.双杂交系统的载体15 Table V.各BD-Matchmaker DNA-BD 载体的比较19 Table VI. RNA起始浓度和PCR扩增循环数之间的关系24 Table VII.单杂交共转化的对照实验的设置29 Table VIII.单杂共转化对照实验:期望的结果29 Table IX.双杂交转化的对照实验的设置33 Table X.双杂交配对筛选的对照实验的设置 Table XI.双杂交共转化的对照实验的设置 Table XII.双杂交共转化的对照实验:期望的结果 Table XIII.用于PCR筛选菌落的Assembling Master Mixs
1 pBait-AbAi载体的构建(酵母报道子的构建) 注:酵母报道子(pBait-AbAi)包含目的顺式作用元件的一个或多个拷贝,且插入到pAbAi载体AbAi r报告基因的上游。大量研究表明最有效的构建应包含目的DNA三个以上的首尾连接的拷贝。首尾连接的拷贝产生方式很多,但对于长度小于20 bp的调控元件,人工合成寡核苷酸是最方便可靠的途径。 (1)设计并合成包含目的序列的两条反向平行的寡核苷酸序列,且两端加上与pAbAi载体酶切产物一致的粘性末端(建议合成一个目的序列的突变序列作为对照,以排除可能的假阳性)。 (2)用TE buffer溶解寡核苷酸至终浓度100 μmol/L。 (3)将正向链和反向链按照1:1的比例混合(退火后的双链寡核苷酸最大浓度为50 μmol/L)。 (4)95 ?C保温30 s,去除二级结构。 (5)72 ?C保温2 min,37 ?C保温2 min,25 ?C保温2min。 注:缓慢退火,有助于双链寡核苷酸的形成。 (6)冰上放置。退火后的产物可贮存在-20 ?C冰箱备用。 (7)酶切1 μL pAbAi载体,用凝胶回收纯化或柱纯化的方式纯化酶切产物。 注:回收前,可用琼脂糖凝胶检测是否酶切完全。 (8)将退火后的寡核苷酸稀释100倍至终浓度为0.5 μmol/L。 (9)在连接反应管中加入如下成分: pAbAi载体(50 ng/μL) 1.0 μL annealed oligonucleotide (0.5 μmol/L) 1.0 μL 10×T4 DNA ligase buffer 1.5 μL BSA(10 mg/mL)0.5 μL Nuclease-free H2O 10.5 μL T4 DNA ligase (400 U/μL)0.5 μL 总体积15 μL 注:如果有必要,可用1 μL nuclease-free H2O代替寡核苷酸作为阴性对照。 (10)将反应体系室温放置连接3 h,转化E coli,采用常规方法检测阳性克隆。
各种SD培养基: 1)SD/-ade(腺嘌呤)/-leu(亮氨酸)/-trp(色氨酸)/-his (组氨酸)(1000 ml)(? “四缺”) 酵母氮源(YNB):6.7g ; -ade/-leu/-trp/-his DO supplement 0.60g (购买来就配好的) ; 葡萄糖20g (即2%) 2)SD/-leu/-trp/-his (1000 ml) 酵母氮源(YNB):6.7g ; -leu/-trp/-his DO supplement 0.62g ; (购买来就配好的) 葡萄糖 20g. (即2%) 3)SD/-leu/-trp (1000 ml) (?“二缺”) 酵母氮源(YNB):6.7g ; -ade/-leu/-trp/-his DO supplement 0.64g (购买来就配好的); 葡萄糖 20g (即2%) 4)SD/-leu (1000 ml) 酵母氮源(YNB):6.7g ; -leu DO supplement 0.69g ; (购买来就配好的) 葡萄糖 20g (即2%) 5)SD/-trp (1000 ml) 酵母氮源(YNB):6.7g ; -ade/-leu/-trp/-his DO supplement 0.74g ; (购买来就配好的) 葡萄糖 20g (即2%) 注意:YNB有两种,一种含有硫酸胺,另外一种不含硫酸胺。我们这用的是含硫酸铵的。(买来就加进去了的)。如果不含硫酸铵,那么要在终浓度0.17%的YNB中再加入0.5%的硫酸铵,即最终在1000 ml溶液中加入总量为6.7g的YNB与硫酸铵。 实际配制的方法是: 1.配制40%的葡萄糖贮存液(贮存在4℃),过滤除菌,待高压灭菌的溶液温度降至55℃ 以下时,再将50ml葡萄糖贮存液加入。(李博士经验这一步不高压,过滤即可使用)2.酵母氮源6.7g,加DO supplement 在920ml水中溶解,调PH至5.8(李博士的经验大 约加10M NaOH 200ul即可),之后补水至950 ml。 3.高压完后待温度降至55℃以下,加入50 ml40%葡萄糖。
(酵母菌储存在-70℃中,引物和质粒DNA储存在-20℃中) 概念: 1. 次序转化:指的是先将一种质粒转化进酵母中(常是DNA-BD/bait plasmid),在选择培养基中选择出阳性克隆,之后再将另外一个质粒(AD fusion library)转化进去。优点:就是比共转化使用更少的质粒DNA,也就是节约质粒DNA。 2. 共同转化:将两种质粒一起转化进酵母中。优点:比次序转化更容易操作。 pGBKT7----的选择物是:kanamycin(卡那霉素)? pGADT7----的选择物是:ampicillin (氨苄西林) ? 各种SD培养基: 1) SD/-ade(腺嘌呤)/-leu(亮氨酸)/-trp(色氨酸)/-his (组氨酸)(1000 ml)(?“四缺”) 酵母氮源(YNB):6.7g ; -ade/-leu/-trp/-his DO supplement 0.60g (购买来就配好的); 葡萄糖 20g (即2%) 2) SD/-leu/-trp/-his (1000 ml) 酵母氮源(YNB):6.7g ; -leu/-trp/-his DO supplement 0.62g ; (购买来就配好的) 葡萄糖 20g. (即2%) 3) SD/-leu/-trp (1000 ml) (?“二缺”) 酵母氮源(YNB):6.7g ; -ade/-leu/-trp/-his DO supplement 0.64g (购买来就配好的); 葡萄糖 20g (即2%) 4) SD/-leu (1000 ml) 酵母氮源(YNB):6.7g ; -leu DO supplement 0.69g ; (购买来就配好的)
酵母单杂交技术 1.酵母单杂交的基本原理 酵母单杂交技术是1993年由酵母双杂交技术发展而来的,其基本原理为:真核生物基因的转录起始需转录因子参与,转录因子通常由一个DNA特异性结合功能域和一个或多个其他调控蛋白相互作用的激活功能域组成,即DNA 结合结构域(DNA—bindingdomain,BD)和转录激活结构域(activationdomain,AD)。用于酵母单杂交系统的酵母GAL4蛋白是一种典型的转录因子,GAL4的DNA结合结构域靠近羧基端,含有几个锌指结构,可激活酵母半乳糖苷酶的上游激活位点(UAS),而转录激活结构域可与RNA聚合酶或转录因子TFIID相互作用,提高RNA聚合酶的活性。在这一过程中,DNA结合结构域和转录激活结构域可完全独立地发挥作用。据此,我们可将GAL4的DNA结合结构域置换为文库蛋白编码基因,只要其表达的蛋白能与目的基因相互作用,同样可通过转录激活结构域激活RNA聚合酶,启动下游报告基因的转录。 酵母单杂交原理示意图 2.酵母单杂交技术的特点 酵母单杂交体系自1993年由Wang和Reed创立以来,在生物学研究领域中已经显示出巨大的威力。应用酵母单杂交体系已经验证了许多已知的DNA与蛋白质之间的相互作用,同时发现了新的DNA与蛋白质的相互作用,并由此找到了多种新的转录因子。近来,已有应用酵母单杂交体系进行疾病诊断的研究报道。随着酵母单杂交体系的不断发展和完善,它在科研、医疗等方面的应用将会越来越广泛。采用酵母单杂交体系能在一个简单实验过程中,识别与DNA特异结合的蛋白质,同时可直接从基因文库中找到编码蛋白的DNA序列,而无需分离纯化蛋白,实验简单易行。由于酵母单杂交体系检测到的与DNA结合的蛋白质是处于自然构象,克服了体外研究时蛋白质通常处于非自然构象的缺点,因而具有很高的灵敏性。目前,多种酵母单杂交体系的试剂盒和相应的cDNA文库已经商品化,为酵母单杂交体系的使用提供了有利的条件。 但酵母单杂交也存在以下缺点:有时由于插入的靶元件与酵母内源转录激活因子可能发生相互作用,或插入的靶元件不需要转录激活因子就可以激活报告基因的转录,因此往往产生假阳性结果。如果酵母表达的AD融合蛋白对细胞有毒性,或融合蛋白在宿主细胞内不能稳定地表达,或融合蛋白发生错误折叠,或者不能定位于酵母细胞核内,以及融合的Gal4AD封闭了蛋白质上与DNA相互作用的位点,则都可能干扰AD融合蛋白结合于靶元件的能力,从而产生假阴性结果。 3.酵母单杂交的基本操作过程
酵母单杂交 酵母单杂交技术最早是1993年由Li等从酵母双杂交技术发展而来,通过对报告基因的表型检测,分析DNA与蛋白之间的相互作用,以研究真核细胞内的基因表达调控。由于酵母单杂交方法检测特定转录因子与顺式作用元件专一性相互作用的敏感性和可靠性,现已被广泛用于克隆细胞中含量微弱的、用生化手段难以纯化的特定转录因子。酵母单杂交(Yeast one-hybrid)是根据DNA结合蛋白(即转录因子)与DNA顺式作用元件结合调控报道基因表达的原理,克隆与靶元件特异结合的转录因子基因(cDNA)的有效方法。其理论基础是:许多真核生物的转录激活子由物理和功能上独立的DNA结合区(DNA-binding domain BD)和转录激活区(Activation domain AD) 组成,因此可构建各种基因与AD的融合表达载体,在酵母中表达为融合蛋白时,根据报道基因的表达情况,便能筛选出与靶元件有特异结合区域的蛋白。理论上,在单杂交检测中,任何靶元件都可被用于筛选一种与之有特异结合区域的蛋白。(百度百科) 酵母单杂交法 酵母单杂交体系(yeast one-hybrid system)常用于研究DNA-蛋白质间的相互作用。 酵母单杂交体系可识别稳定结合于DNA上的蛋白质,可在酵母细胞内研究真核DNA-蛋白质间的相互作用,并通过筛选DNA文库直接获得靶序列相互作用蛋白的编码基因。也可用于分析鉴定细胞中转录调控因子与顺式作用元件相互作用。 酵母单杂交原理:将已知的顺式作用元件构建到最基本启动子(minimal promoter,Pmin)上游,把报告基因连接到Pmin下游。将待测转录因子的cDNA与酵母转录激活结构域(activation domain,AD)融合表达载体导入细胞,该基因产物如果能够与顺式作用元件结合,而激活Pmin启动子使报告基因表达。 酵母单杂交体系主要用于分离编码结合于特定顺式调控元件或其他DNA位点的功能蛋白编码基因,验证反式转录调控因子的DNA结合结构域,准确定位参与特定蛋白质结合的核苷酸序列。
Two-hybrid assay of yeast transformation Qi's protocol (This method can be used for both laboratory and industrial yeast) Yeast strain we used for two-hybrid is S. cerevisiae HF7c (MATa ura3-52 his3-200 ade2-101 lys2-801 trp1-901 leu2-3,112 gal4-542 gal80-538 LYS2::GAL1UAS-GAL1TATA-HIS3 URA3::GAL4 17mers(x3)-CyC1TATA-LacZ), which contains the two reporter genes LacZ and HIS3, was used in two-hybrid analysis. Yeast can be co-transformation with plasmids carrying the different GAL4 DNA binding domain-target protein fusions (TRP1marker) and plasmid carrying the GAL4 activation domain (LEU2marker) to perform the protein-protein interaction study or two-hybrid screening. The transformation method is modified from Schiestl and Gietz (1989) GAL4 DNA binding domain vector (pGBT8 or pGBT9, difference only in polylinker, relatively low expression, TRP1 marker) GAL4 activation domain vector (pGAD424, low expression; pGADGH, high expression, LEU2 marker ) Transformation protocol for unique plasmid or for two known plasmids 1. From the stock (-70aC) the yeast were inoculated on YPAD plate for 2 days at 28-32aC 2. Inoculate 2-3 independent yeast colonies to 10 ml YPAD medium for the preinoculation, shaking at 28-32aC for 10 to 12 hours, check OD600. 3. Make an inoculation of the volume necessary for the transformation. (Usually 10 ml of OD600 0.3 cells or 5 ml of OD600 0.8 is used for each sample transformation. You can use the formula: OD -------- 2n ------------- x Vinoc. = Vpreinoc. ODpre OD is OD600 of cells you will use for transformation, 0.3-0.8 of OD600 is good for LiOAc transformation. ODpre is OD600 of the preinoculation. n is the generation time of yeast, different yeast strain has different generation time. The generation time for HF7C is 1.5 hours in rich medium and 2 hours in minimal medium. Vinoc. The volume you need for the transformation. Vpreinoc. is the volume you need take from preinoculation. Shaking at 28-32aC and an overnight inoculation is recommended
模块七蛋白质之间的相互作用 1.实验目的 本实验以重组质粒和酵母细胞为材料, 学习检测蛋白质相互作用的基本原理和 技术方法。主要介绍酵母双杂交的基本原理与操作技术 ; 让学生了解和掌握酵母双 杂交系统的应用 ; 掌握酵母感受态的制备的基本原理和主要的操作步骤。 2.实验原理 1989 年Fields 和Song 等人根据当时人们对真核生物转录起始过程调控的认 识(即细胞内基因转录的起始需要转录激活因子的参与,提出并建立了酵母双杂交系 统。该系统作为发现和研究活细胞体内的蛋白质与蛋白质之间的相互作用的技术平 台 ,近几年得到了广泛的运用和发展。 相比于其它蛋白质筛选系统,酵母双杂交系统具有以下优点:(1 检测在真核活细 胞内进行 ,在一定程度上代表细胞内的真实情况。(2 作用信号是在融合基因表达后, 在细胞内重建转录因子的作用而给出的 ,省去了纯化蛋白质的繁琐步骤。(3 检测结果是基因表达产物的积累效应,因而可检测存在于蛋白质之间的微弱或暂时的相互 作用。(4 酵母双杂交系统可采用不同组织、器官、细胞类型和分化时期材料构建cDNA 文库 ,能分析细胞质、细胞核及膜结合蛋白等多种不同亚细胞部位及功能蛋白。(5 通过mRNA 产生多种稳定的酶使信号放大。同时,酵母表型、X-Gal 及 HIS3 蛋白表达等检测方法均很敏感。 酵母双杂交系统也具有一定的局限性。首先 , 经典的双杂交系统分析蛋白间的 相互作用定位于细胞核内,因而限制了该系统对某些细胞外蛋白和细胞膜受体蛋白 的研究。酵母双杂交系统的另一个局限性是“假阳性”。在酵母双杂交系统建立的 初期阶段 ,由于仅仅采用β-半乳糖苷酶这一单一的报告基因体系,这种报告基因的表 达往往不能十分严谨地被控制,因此容易产生假阳性。由于某些蛋白本身具有激活 转录的功能或在酵母中表达时发挥转录激活作用, 使DNA 结合结构域融合蛋白在 无特异激活结构域的情况下也可被激活转录。另外某些蛋白表面含有对多种蛋白
4月4日划线配培养基TE/LIAC PEG/LIAC 配置培养基(YPD YPDA)取酵母细胞划线30°生长3天。 需要用品:三角瓶灭菌封口膜酵母提取物蛋白胨 注:以下所有涉及菌的操作均需在超净台中完成。 4月6号星期三 (1)选择2-3mm的单克隆(枪头吸取)放入3-5ml的YPDA液体培养基,30°摇菌200rpm,8h 7号下午开始,过夜培养,次日若菌液浓度达到标准,可先置于4度冰箱保存。 需要用品:200ul灭菌枪头、50ml三角瓶、YPDA液体培养基、摇床。 4月7号星期四 (2)吸取2.5-10ul酵母培养液,加入25mlYPDA液体培养基,摇菌16-20h直到OD值0.15-0.3。 下午4点开始8号8点结束 Tips:由于第一次活化的菌夜浓度不一,此处建议设置梯度,分别取2.5、5、10 ul酵母培养液,加入25ml YPDA液体培养基(转化5个以下质粒的话,25ml菌量就够后续使用)。 4月8号星期五 (3)将菌液转移至灭菌的50ml离心管中,用天平配平后,室温下700g离心5分钟。 (4)弃掉上清,加入50ml新鲜的YPDA液体重悬菌体(由于离心转速较低,沉淀易悬起来,故倒掉上清液时要小心操作)。 (5)30°震荡培养,直到OD值达到0.4-0.5 (3-5h)。8号8点开始下午一点结束进行以下操作之前,配置好TE/LiAc溶液,并准备好冰浴。 (6)将上述菌液转移至一个灭菌的50ml离心管中,用天平配平后,室温下700g离心5分钟。(7)弃掉上清,用30ml无菌水重悬菌体(小心操作)。 (8)再次用天平配平后,室温下700g离心5分钟,弃去上清,加入1.5ml 1.1xTE/LIAC重悬菌体。(9)将上述溶液转移到灭菌的1.5mlEP管中,高速离心15s。 (10)弃去上清,加入600ul 1.1x TE/LIAC,感受态细胞制备完成,置于冰上待用。 需要物品:50ml灭菌离心管、50ml 三角瓶、1.5ml EP管、5ml灭菌枪头、1ml灭菌枪头、灭菌ddH2O、YPDA液体培养基、1.1x TE/LIAC。 1.1x TE/LIAC10ML 10xTE 1.1ml 10xliac 1.1ml Dh2O8.8ml
4月4日划线配培养基 TE/LIAC PEG/LIAC 配置培养基(YPD YPDA)取酵母细胞划线 30°生长3天。 需要用品:三角瓶灭菌封口膜酵母提取物蛋白胨 注:以下所有涉及菌的操作均需在超净台中完成。 4月6号星期三 (1)选择2-3mm的单克隆(枪头吸取)放入3-5ml的YPDA液体培养基,30°摇菌200rpm,8h 7号下午开始,过夜培养,次日若菌液浓度达到标准,可先置于4度冰箱保存。 需要用品:200ul灭菌枪头、50ml三角瓶、YPDA液体培养基、摇床。 4月7号星期四 (2)吸取2.5-10ul酵母培养液,加入25ml YPDA液体培养基,摇菌16-20h直到OD值0.15-0.3。 下午4点开始 8号 8点结束 Tips:由于第一次活化的菌夜浓度不一,此处建议设置梯度,分别取2.5、5、10 ul酵母培养液,加入25ml YPDA液体培养基(转化5个以下质粒的话,25ml菌量就够后续使用)。 4月8号星期五 (3)将菌液转移至灭菌的50ml离心管中,用天平配平后,室温下700g离心5分钟。 (4)弃掉上清,加入50ml新鲜的YPDA液体重悬菌体(由于离心转速较低,沉淀易悬起来,故倒掉上清液时要小心操作)。 (5)30°震荡培养,直到OD值达到0.4-0.5 (3-5h)。8号 8点开始下午一点结束进行以下操作之前,配置好TE/LiAc溶液,并准备好冰浴。 (6)将上述菌液转移至一个灭菌的50ml离心管中,用天平配平后,室温下700g离心5分钟。(7)弃掉上清,用30ml无菌水重悬菌体(小心操作)。 (8)再次用天平配平后,室温下700g离心5分钟,弃去上清,加入1.5ml 1.1xTE/LIAC重悬菌体。(9)将上述溶液转移到灭菌的1.5ml EP管中,高速离心15s。 (10)弃去上清,加入600ul 1.1x TE/LIAC,感受态细胞制备完成,置于冰上待用。 需要物品:50ml 灭菌离心管、50ml 三角瓶、1.5ml EP管、5ml灭菌枪头、1ml灭菌枪头、灭菌ddH2O、YPDA液体培养基、1.1x TE/LIAC。 1.1x TE/LIAC 10ML 10xTE 1.1ml 10xliac 1.1ml Dh2O 8.8ml
蛋白的酵母双杂交实验 ——以钓饵蛋白筛选cDNA 文库研究蛋白相互作用 第一部分 系统简介 1. 实验原理 蛋白的酵母双杂交实验是以酵母的遗传分析为基础,研究反式作用因子之间的相互作用 对真核基因转录调控影响的实验。很早就已知道,转录活化蛋白可以和DNA 上特异的序列结合而启动相应基因的转录反应。这种DNA 结合与转录激活的功能是由转录活化蛋白上两个相互独立的结构域即DNA 结合结构域(Binding Domain, BD)和转录活化结构域(Activation Domain, AD)分别来完成的,并且这两个结构域对于基因的转录活化都是必须的。目前酵母双杂交实验采用的系统有LexA 系统和Gal4系统两种。在LexA 系统中,DNA 结合结构域由一个完整的原核蛋白LexA 构成,转录活化结构域则由一个88个氨基酸的酸性的大肠杆菌多肽B42构成,它在酵母中可以活化基因的转录; 在 Gal4系统中,BD 和AD 分别由Gal4蛋白上不同的两个结构域(1-147aa 与768-881aa)构成。在利用GAL4系统筛选cDNA 文库或研究蛋白间的相互作用时,DNA 结合结构域与靶蛋白即“诱饵”相结合,转录活化结构域与文库蛋白或要验证的蛋白相结合。一般情况下,单独的BD 可以与GAL4上游活化序列(GAL UAS )结合但不能引起转录,单独的AD 则不能与GAL UAS 结合,只有当BD 与AD 分别表达的融合蛋白由于相互作用而导致两者在空间上相互靠近时,BD 与AD 才能与GAL UAS 结合并且引起报道基因的转录。在BD 与AD 要导入的酵母菌AH109中,通过基因工程的方法在GAL4 UASs 和启动子的下游构建了3个报道基因——ADE2,HIS3,MEL1(或LacZ ),因此可以通过营养缺陷筛选和酵母菌表型的改变来筛选或验证两个蛋白之间是否存在相互作用。GAL4系统的原理如图所示: 图一:酵母双杂交系统工作原理 Kan r Amp r pGBKT7-bait pACT2-cDNA
模块七蛋白质之间的相互作用 1. 实验目的 本实验以重组质粒和酵母细胞为材料,学习检测蛋白质相互作用的基本原理和技术方法。主要介绍酵母双杂交的基本原理与操作技术;让学生了解和掌握酵母双杂交系统的应用;掌握酵母感受态的制备的基本原理和主要的操作步骤。 2. 实验原理 1989年Fields和Song等人根据当时人们对真核生物转录起始过程调控的认识(即细胞内基因转录的起始需要转录激活因子的参与),提出并建立了酵母双杂交系统。该系统作为发现和研究活细胞体内的蛋白质与蛋白质之间的相互作用的技术平台,近几年得到了广泛的运用和发展。 相比于其它蛋白质筛选系统,酵母双杂交系统具有以下优点:(1)检测在真核活细胞内进行,在一定程度上代表细胞内的真实情况。(2)作用信号是在融合基因表达后,在细胞内重建转录因子的作用而给出的,省去了纯化蛋白质的繁琐步骤。(3)检测结果是基因表达产物的积累效应,因而可检测存在于蛋白质之间的微弱或暂时的相互作用。(4)酵母双杂交系统可采用不同组织、器官、细胞类型和分化时期材料构建cDNA文库,能分析细胞质、细胞核及膜结合蛋白等多种不同亚细胞部位及功能蛋白。(5)通过mRNA产生多种稳定的酶使信号放大。同时,酵母表型、X-Gal 及HIS3 蛋白表达等检测方法均很敏感。 酵母双杂交系统也具有一定的局限性。首先,经典的双杂交系统分析蛋白间的相互作用定位于细胞核内,因而限制了该系统对某些细胞外蛋白和细胞膜受体蛋白的研究。酵母双杂交系统的另一个局限性是“假阳性”。在酵母双杂交系统建立的初期阶段,由于仅仅采用β-半乳糖苷酶这一单一的报告基因体系,这种报告基因的表达往往不能十分严谨地被控制,因此容易产生假阳性。由于某些蛋白本身具有激活转录的功能或在酵母中表达时发挥转录激活作用,使DNA结合结构域融合蛋白在无特异激活结构域的情况下也可被激活转录。另外某些蛋白表面含有对多种蛋白质的低亲和力区域,能与其他蛋白形成稳定的复合物,从而引起报告基因的表达,产生“假阳性”结果。产生“假阴性”结果的原因可能有许多蛋白质间的相互作用依赖于翻译后加工如糖基化、磷酸化和二硫键形成,还有些蛋白的正确折叠和功能有赖于某些非酵母蛋白的辅助等。 现在的酵母双杂交系统大都采用多种报告基因,如AH109酵母株含有三类报告基因—ADE2、HIS3、MEL1/lacZ,这三类报告基因受控于三种完全不同、异源性的GAL4-反应元件和三类启动子元件-GAL1、GAL2以及MEL1(如图6-1-1)。通过这种方法就消除了两类最主要的假阳性,一类是融合蛋白可以直接与GAL4结合位点结合或者是在结合位点附近结合所带来的假阳性;另一类是融合蛋白和某种转录因子结合后再结合到特定的TA TA盒上所带来的假阳性。ADE2一种报告基因就已经能够提供较强的营养选择压力,这时选择性地使用HIS3报告基因,一来可以降低假阳性率;二来可以控制筛选的严格性(如果需要筛选与诱饵蛋白具有较强结合的蛋白,就可以同时使用ADE2、HIS3两种报告基因;如果只需要筛选与诱饵蛋白具有中等强度或较弱结合的蛋白,就可以使用ADE2或HIS3两者中的一种)。MEL1和lacZ分别编码α-半乳糖苷酶和β-半乳糖苷酶,可以作用于相应的底物
酵母单杂交技术(Yeast One-hybrid method) 一、原理 酵母单杂交方法是根据DNA结合蛋白(即转录因子)与DNA顺式作用元件结合调控报道基因表达的原理来克隆编码目的转录因子的基因(cDNA)。该方法也是在细胞内(in vivo)分析鉴定转录因子与顺式作用元件结合的有效方法。将已知的特定顺式作用元件构建到最基本启动子(minimal promoter,Pmin)上游,Pmin启动子下游连接报道基因。进行c DNA 融合表达文库筛选时,编码目的转录因子的c DNA 融合表达载体被转化进入酵母细胞后,其编码产物(转录因子)与顺式作用元件结合,就可以激活Pmin启动子,并促使报道基因表达。根据报道基因的表达,筛选出与已知顺式元件结合的转录因子。 二、实验步骤 ①构建报道子载体:将已知的特定顺式作用元件按同一方向随机串联到一个最小启动子(minimal promoter,Pmin)上游,其下游连有报道基因; ②将构建好的报道子载体转化酵母细胞筛选合适的3-AT浓度; ③提取总RNA,合成c DNA双链; ④将报道子、c DNA和融合表达载体共转化到酵母中,在相应的缺陷性SD培养基上进行筛选阳性克隆; ⑤对阳性克隆进行鉴定,去除假阳性克隆; ⑥对所获得的阳性克隆进行测序,为进一步分析打下基础。如可进一步分析DNA确切的结合位点。 三、图片分析 1、选择YM4271[ pHISi3NF4] 克隆株重复进行3-AT 梯度试验,在没有3-AT的SD/-His平板上生长状态很好,在15mM 3-AT存在下被明显抑制,而在30mM 3-AT存在下完全不能生长。故选择15mM为筛库实验的3-AT工作浓度。 2、经9天培养,在SD/-Leu/-His/ +15mM[3-AT]选择培养基上共长出351个大小不同的阳性候选克隆。取100个菌落划线于30mM 3-AT和60mM[3-AT]平板,仅有1个克隆被淘汰。证实15mM[3-AT]的选择是正确的。挑取阳性候选克隆菌落提取酵母质粒并转化E .coli Top 10F'后,酶切鉴定,共得到31个初筛阳性克隆。 四、严谨性分析 1、为了克服His3基因的渗漏表达对筛库实验结果的影响,我们选择克隆株重复进行3-AT 梯度试验。菌株在没有3-AT的SD/-His平板上生长状态很好,在15mM 3-AT下被明显抑制,
第四章基因文库构建及酵母双杂交技术 1 基因组文库的构建 基因组文库(genomic library)将某种生物细胞的整个基因组DNA切割成大小合适的片断,并将所有这些片断都与适当的载体连接,引入相应的宿主细胞中保存和扩增。 理论上讲,这些重组载体上带有了该生物体的全部基因,称为基因文库。 1. 1构建基因文库的载体选用 载体能够容载的DNA片断大小直接影响到构建完整的基因文库所需要的重组子的数目。
第四章基因文库构建及酵母双杂交技术1.1.1对载体的要求:载体容量越大,所要求的DNA片断数目越少,所需的重组子越少。 1.1.2目前常用的载体: 载体系列(容量为24 kp )、cosmid载体(容量为50 kb )、YAC(容量为1 Mb )、BAC (容量为300 kb) 1.2 基因文库构建的一般步骤 1.2.1染色体DNA大片段的制备:断点完全随机,片断长度合适于载体连接。不能用一般的限制性内切酶消化法,使用物理切割法或不完全酶切法。 1.2.2载体与基因组DNA大片段的连接:直接连接、人工接头或同聚物加尾。
噬菌体载体构建基因组文库
第四章基因文库构建及酵母双杂交技术2 cDNA文库的构建 cDNA克隆的基本过程是通过一系列,酶酶催作用,使poly(A) mRNA转变成双链cDNA群体并插入到适当的载体分子上,转化大肠杆菌寄主细胞,构建包含所有基因编码序列的cDNA基因文库。 2.1高质量mRNA的制备 应用Promega PolyAT tract mRNA Isolation System 分离Poly(A)RNA。将Biotinylated Oligo(dT)引物与细胞总RNA共温育,加入与微磁球相连的Streptavidin,用磁场吸附与PMP相连的SA-Biotinylated Oligo(dT)-mRNA。
酵母单杂交(yeast one hybrid)技术 (2013-03-23 05:24:00) 转载▼ 分类:分子生物学专业 标签: 酵母单杂交 gal4蛋白 酵母双杂交技术 报告基因 文化 酵母单杂交(yeast one hybrid)技术,是体外分析DNA与细胞内蛋白质相互作用的一种方法,通过对酵母细胞内报告基因表达状况的分析,来鉴别DNA结合位点并发现潜在的结合蛋白基因,或对DNA结合位点进行分析。 运用此技术,能筛选到与DNA结合的蛋白质,并可直接从基因文库中得到编码该蛋白质的核苷酸序列,而无需复杂的蛋白质分离纯化操作,故在蛋白研究中,具有一定的优势;而且,酵母属真核细胞,通过酵母系统得到的结果,比其他体外技术获得的结果,更能体现真核细胞内基因表达调控的真实情况。 1 酵母单杂交技术的原理 酵母单杂交技术,最早是1993年由Li et al从酵母双杂交技术发展而来,酵母双杂交技术通过对报告基因的表型进行检测以实现对蛋白质间相互作用的研究,而酵母单杂交技术则通过对报告基因的表型检测,分析DNA、蛋白之间的相互作用,以研究真核细胞内的基因表达调控。 目前认为真核生物的转录起始,需要转录因子的参与。这些转录因子通常由一个DNA 特异性结合功能域和一个或多个与其他调控蛋白相互作用的激活功能域组成,即DNA结合结构域(DNA-binding domain, BD)和转录激活结构域(activation domain, AD)。 用于酵母单杂交系统的酵母GAL4蛋白即是一种典型的转录因子。 研究表明GAL4的DNA结合结构域,靠近羧基端,含有几个锌指结构,可激活酵母半乳糖苷酶的上游激活位点(UAS);而转录激活结构域,可与RNA聚合酶或转录因子TFIID相互作用,提高RNA聚合酶的活性。 在这一过程中,DNA结合结构域和转录激活结构域可完全独立地发挥作用。据此,我们可将GAL4的DNA结合结构域置换为其他蛋白,只要它能与我们想要了解的目的基因相互作用,就照样可以通过其转录激活结构域激活RNA聚合酶,从而启动对下游报告基因的转录。 正是基于这一理论,酵母单杂交系统由2部分组成: (1)将文库蛋白片段,与GAL4转录激活域融合表达的cDNA文库质粒; (2)含有目的基因和下游报告基因的报告质粒。
LexA酵母双杂交系统简介 一、LexA酵母双杂交系统的设计原理 报告质粒p8op-LacZ的GAL4 UAS编码序列被完全去除,因此在缺乏LexA融合激活剂的情况下,报告基因LacZ的转录活性为零,该基因的筛选标志为URA3,可以作为有自主复制能力的质粒存在于酵母EGY48菌株中,也可以被整合到EGY48基因组DNA上。 质粒pLexA的筛选标志为HIS3,在双杂交系统中用于表达DNA-BD(202个氨基酸残基组成的LexA蛋白)与目标蛋白(钓饵,Bait)的融合蛋白,该融合体的表达受酵母强启动子ADH1的调控,选择与报告基因的操纵子LexA×8结合。 质粒pB42AD的筛选标志为TRP1,在其供外源基因插入的多克隆位点(EcoR I与Xho I)上游,含有SV40核定位(SV40 nuclear localization)、HA(血凝素)及AD(来自于E.coli的88个氨基酸残基组成的B42蛋白)等几种编码序列,共同组成可以启动报告基因转录表达的激活成份。在酵母EGY48的基因组中还整合有另一个报告基因Leu,它与LacZ报告基因具有相同的操纵子-LexA,但两者启动子不同。 根据双杂交系统的原理,如果某一复合物同时具有DNA-BD和AD的活性,即可激活报告基因的转录和表达。分别将待测蛋白X、Y的编码序列插入pLexA质粒载体和pB42AD质粒载体的多克隆位点中,然后共同转入含有报告基因的酵母菌株,如果蛋白X与Y能相互作用,则启动报告基因的转录和表达,通过检测报告基因的表达情况,就可以间接反映蛋白X、Y是否具有相互作用以及作用的强弱。 如果将蛋白Y换为取自组织或血液的cDNA文库,则可用X从该文库中筛选出能与其相互作用的蛋白,并且可以获得编码这些蛋白的cDNA。 二、商品化酵母双杂交系统的组成 1. 载体质粒:pLexA、pB42AD、p8op-LacZ、pB42AD-DNA文库 2. 酵母菌株:EGY48、EGY48(p8op-LacZ)、YM4271(EGY48的伴侣菌株) 3. 大肠杆菌菌株:E.coli KC8株 4. 对照质粒: 质粒用途 pLexA-53,pB42AD-T 阳性对照 pLexA-Pos(LexA/GAL4 AD融合蛋白〕阳性对照 pLexA-Lam(LaminC蛋白少与其它蛋白相互作用) 假阳性检测质粒 5. 引物: pLexA测序引物及pB42AD测序引物。 三、酵母双杂交实验的基本流程 1. 将报告基因p8op-LacZ转化酵母EGY48菌株,用培养基SD/-Ura筛选。 2. 同时构建或扩增DNA文库,并纯化足够的质粒以转化酵母细胞。 3. 构建DNA-BD/靶蛋白质粒pLexA-X,作为钓饵(bait)。 4. 将上述钓饵质粒pLexA-X转化EGY48(p8op-LacZ)细胞株,用SD/-His/-Ura筛选;并用固体诱导培养基SD/Gal/Raf/-His/-Ura检测此DNA-BD/靶蛋白是否具有直接激活报告基因的活性,以及对酵母细胞是否具有杀伤毒性。 转化质粒选择培养基克隆生长情况说明