
Comparisons of O-acylation and Friedel e Crafts acylation of phenols and acyl chlorides and Fries rearrangement of phenyl esters in tri ?uoromethanesulfonic acid:effective synthesis of optically active homotyrosines
Ryo Murashige a ,b ,Yuka Hayashi a ,Syo Ohmori a ,Ayuko Torii a ,Yoko Aizu a ,Yasuyuki Muto a ,Yuta Murai c ,Yuji Oda a ,Makoto Hashimoto c ,*
a
Department of Agricultural and Life Science,Obihiro University of Agriculture and Veterinary Medicine,Inada-cho,Obihiro 080-8555,Hokkaido,Japan b
Department of Pharmaceutical Sciences,Himeji Dokkyo University,7-2-1Kamioono,Himeji 670-8524,Hyogo,Japan c
Division of Applied Science,Graduate School of Agriculture,Hokkaido University,Kita 9,Nishi 9,Kita-ku,Sapporo 060-8589,Japan
a r t i c l e i n f o
Article history:
Received 14September 2010
Received in revised form 9November 2010Accepted 10November 2010
Available online 16November 2010Keywords:
Friedel e Crafts acylation Fries rearrangement O-Acylation
Tri ?uoromethanesulfonic acid Homotyrosine
a b s t r a c t
Reactions involving phenol derivatives and acyl chlorides have to be controlled for competitive O-ac-ylations and C-acylations (Friedel e Crafts acylations and Fries rearrangements)in acidic condition.The extent for these reactions in tri ?uoromethanesulfonic acid (TfOH),which is used as catalyst and solvent,is examined.Although diluted TfOH was needed for effective O-acylation,concentrated TfOH was re-quired for effective C-acylations in mild condition.These results have been applied to the novel synthesis of homotyrosine derivatives.Both Fries rearrangement of N -TFA e Asp(OBn)e OMe and Friedel e Crafts acylation of phenol with N -TFA e Asp(Cl)e OMe in TfOH afforded the homotyrosine skeleton,followed by reduction and deprotection afforded homotyrosines maintaining stereochemistry of Asp as an optically pure form.
ó2010Elsevier Ltd.All rights reserved.
1.Introduction
Hydroxyaryl ketones are versatile intermediates in the synthesis of biologically active compounds and both Friedel e Crafts acylation of phenol derivatives and Fries rearrangement of acyloxy benzenes are major pathways for their preparations.1These reactions have been promoted with Lewis acid.Superacidic systems that may be formed by mixing appropriate Lewis and Br?nsted acids have been the subject of much academic and industrial research.2Tri-?uoromethanesulfonic acid (TfOH),which can be considered a su-per-acid,forms water-stable salts with non-hydrolyzable metals.3The rare-earth metal tri ?ates are also useful for the purpose,4but it is sometimes dif ?cult to completely archive these reactions.We recently reported that neat TfOH catalyzed Friedel e Crafts acylation.This involved stoichiometric amounts of non-phenolic aromatics and acid chlorides at a side chain of aspartic and glutamic acid whose stereochemistry was maintained in a very mild condition.5In the reaction,TfOH was used as a catalyst of Friedel e Crafts acylation and as a solvent for amino acid derivatives.For the phenol derivatives,it is necessary to take into account that the O-acylation and C-
acylation (Friedel e Crafts acylation and Fries rearrangement)are attractive objects for synthetic chemistry (Scheme 1).The estab-lishment of comparisons of these reactions is necessary for applied to derivatizations of phenols used for amino acid modi ?cations.
Homotyrosine (hTyr),as a nonproteinogenic a -amino acid,elongates methylene in a side chain of tyrosine (Tyr).The hTyr is present as a component of diverse natural products that have im-portant biological activities 6and its derivatives are important precursors for the total synthesis of some natural products.7Also
Scheme 1.Schematic relationships of the reactions of phenols and acyl chlorides,and O -acyloxy benzenes in TfOH.
*Corresponding author.Tel./fax:t81117063849;e-mail address:hasimoto@abs.agr.hokudai.ac.jp (M.Hashimoto).
Contents lists available at ScienceDirect
Tetrahedron
journal h omepage:ww w.el https://www.doczj.com/doc/49190867.html,/locate/tet
0040-4020/$e see front matter ó2010Elsevier Ltd.All rights reserved.doi:10.1016/j.tet.2010.11.047
Tetrahedron 67(2011)641e 649
sometimes it produces different biological activities when replac-ing Tyr with hTyr in bioactive peptides.8
Asymmetric and ef?cient synthesis of hTyr is important.There are several reports of the synthesis of hTyr;for example,synthetic methods include:enzymatic resolution,6,9Suzuki-coupling,10dia-stereoselective Michael addition,11and catalytic asymmetric hydro-genation.12These methods require special reagents or precursors and phenolic hydroxyl groups would not be expected to tolerate the synthesis conditions.
Furthermore,these methods require the preparation of special reagents or precursors for the asymmetric synthesis of both en-antiomers.Few reports are available,involving the synthetic routes without phenolic hydroxyl protection.13
Amino acids are one of the most popular precursors for ster-eocontrolled synthesis and are easily available for asymmetric synthesis.14The Friedel e Crafts reactions between phenol and a side chain of aspartic acid(Asp)is one of the key reactions for synthesis of optically pure hTyr enantiomers’skeletons.To our best knowl-edge,however,there are no studies for Friedel e Crafts between phenols and Asp derivatives.Because the a-amino acid equivalents are insoluble to these organic solvents in the presence of Lewis acid, the reaction mixture forms a suspension.
In this paper,we report the comparisons of O-and C-acylation for the phenol reaction with acyl halides,and Fries rearrangement of phenyl ester using TfOH.Furthermore,the established conditions were applied to effective synthesis of optically pure hTyr using stoi-chiometric amounts of unprotected phenol and acyl chloride de-rivative of Asp.
2.Results and discussion
2.1.O-Acylation of phenol derivatives and acyl chlorides in
the presence of TfOH
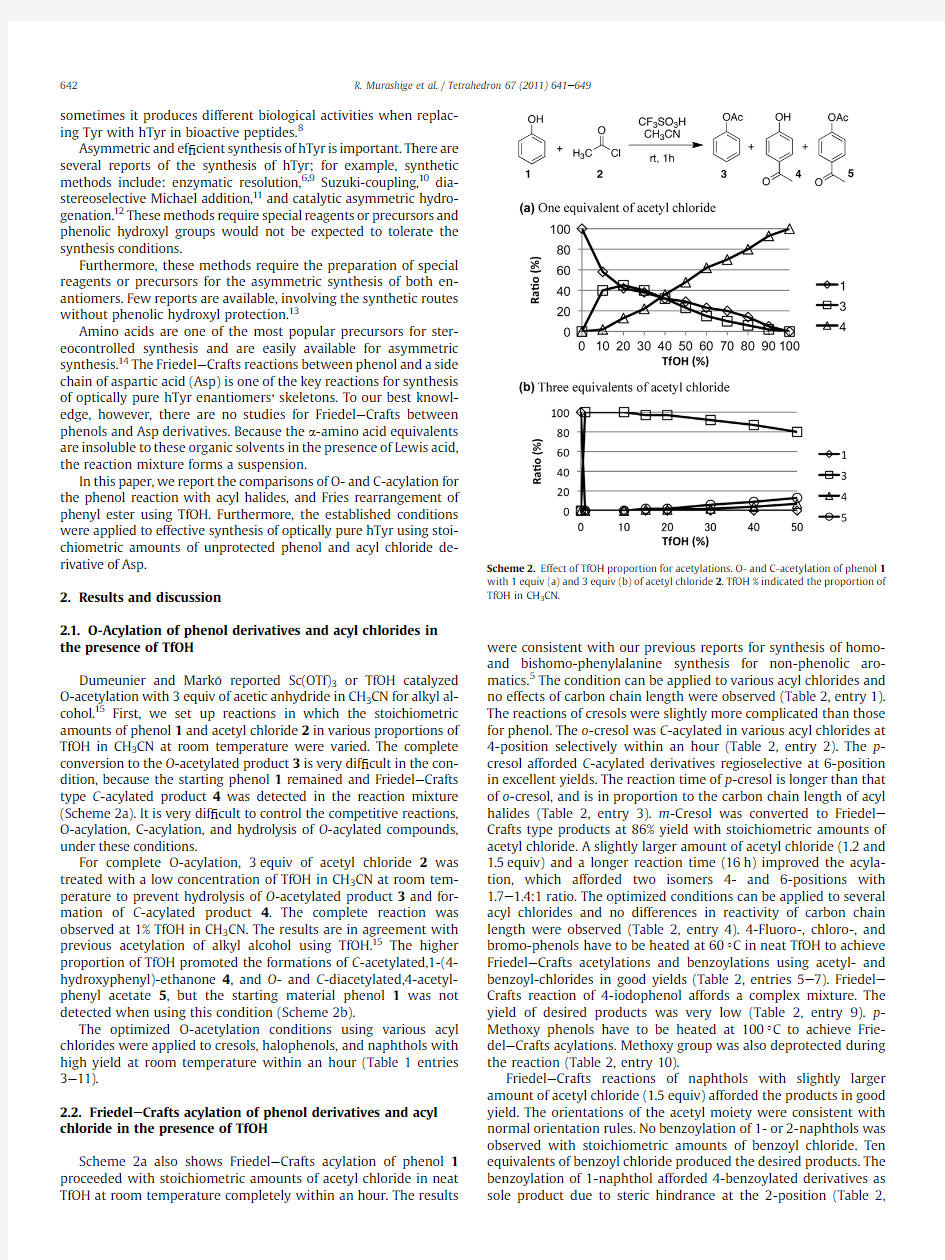
Dumeunier and Mark o reported Sc(OTf)3or TfOH catalyzed O-acetylation with3equiv of acetic anhydride in CH3CN for alkyl al-cohol.15First,we set up reactions in which the stoichiometric amounts of phenol1and acetyl chloride2in various proportions of TfOH in CH3CN at room temperature were varied.The complete conversion to the O-acetylated product3is very dif?cult in the con-dition,because the starting phenol1remained and Friedel e Crafts type C-acylated product4was detected in the reaction mixture (Scheme2a).It is very dif?cult to control the competitive reactions, O-acylation,C-acylation,and hydrolysis of O-acylated compounds, under these conditions.
For complete O-acylation,3equiv of acetyl chloride2was treated with a low concentration of TfOH in CH3CN at room tem-perature to prevent hydrolysis of O-acetylated product3and for-mation of C-acylated product4.The complete reaction was observed at1%TfOH in CH3CN.The results are in agreement with previous acetylation of alkyl alcohol using TfOH.15The higher proportion of TfOH promoted the formations of C-acetylated,1-(4-hydroxyphenyl)-ethanone4,and O-and C-diacetylated,4-acetyl-phenyl acetate5,but the starting material phenol1was not detected when using this condition(Scheme2b).
The optimized O-acetylation conditions using various acyl chlorides were applied to cresols,halophenols,and naphthols with high yield at room temperature within an hour(Table1entries 3e11).
2.2.Friedel e Crafts acylation of phenol derivatives and acyl chloride in the presence of TfOH
Scheme2a also shows Friedel e Crafts acylation of phenol1 proceeded with stoichiometric amounts of acetyl chloride in neat TfOH at room temperature completely within an hour.The results were consistent with our previous reports for synthesis of homo-and bishomo-phenylalanine synthesis for non-phenolic aro-matics.5The condition can be applied to various acyl chlorides and no effects of carbon chain length were observed(Table2,entry1). The reactions of cresols were slightly more complicated than those for phenol.The o-cresol was C-acylated in various acyl chlorides at 4-position selectively within an hour(Table2,entry2).The p-cresol afforded C-acylated derivatives regioselective at6-position in excellent yields.The reaction time of p-cresol is longer than that of o-cresol,and is in proportion to the carbon chain length of acyl halides(Table2,entry3).m-Cresol was converted to Friedel e Crafts type products at86%yield with stoichiometric amounts of acetyl chloride.A slightly larger amount of acetyl chloride(1.2and 1.5equiv)and a longer reaction time(16h)improved the acyla-tion,which afforded two isomers4-and6-positions with 1.7e1.4:1ratio.The optimized conditions can be applied to several acyl chlorides and no differences in reactivity of carbon chain length were observed(Table2,entry4).4-Fluoro-,chloro-,and bromo-phenols have to be heated at60 C in neat TfOH to achieve Friedel e Crafts acetylations and benzoylations using acetyl-and benzoyl-chlorides in good yields(Table2,entries5e7).Friedel e Crafts reaction of4-iodophenol affords a complex mixture.The yield of desired products was very low(Table2,entry9).p-Methoxy phenols have to be heated at100 C to achieve Frie-del e Crafts acylations.Methoxy group was also deprotected during the reaction(Table2,entry10).
Friedel e Crafts reactions of naphthols with slightly larger amount of acetyl chloride(1.5equiv)afforded the products in good yield.The orientations of the acetyl moiety were consistent with normal orientation rules.No benzoylation of1-or2-naphthols was observed with stoichiometric amounts of benzoyl chloride.Ten equivalents of benzoyl chloride produced the desired products.The benzoylation of1-naphthol afforded4-benzoylated derivatives as sole product due to steric hindrance at the2-position(Table2
, Scheme2.Effect of TfOH proportion for acetylations.O-and C-acetylation of phenol1 with1equiv(a)and3equiv(b)of acetyl chloride2.TfOH%indicated the proportion of TfOH in CH3CN.
R.Murashige et al./Tetrahedron67(2011)641e649 642
entry 10).On the other hand,2-naphthol afforded 1-and 3-ben-zoylated products (20:1)under these conditions (Table 2,entry 11).
2.3.Fries rearrangements of O -acyloxy benzenes
The reactions involving Fries rearrangements of O -acyloxy benzenes with rare-earth metal tri ?ates have been out under heating conditions.16There have been reports that combinations of rare metal tri ?ates or phosphorous oxychloride in the presence of methanesulfonic acid were effective for Fries rearrangement,but the reaction was also carried out under heating conditions.17The phenyl acetate 3was subjected in various proportions of TfOH in CH 3CN at room temperature for an hour.Both Fries rearrangement and hydrolysis of the starting material 3were observed with less than 60%TfOH in CH 3CN.
The phenyl acetate 3was completely rearranged to p -acetyl-phenol 4in neat TfOH.No o -acetylphenol and 4-acetylphenyl
acetate 5were observed during the reaction (Scheme 3and Table 3,entry 1).
The rearrangement of o -acyloxy toluenes afforded regiospeci ?c products,in which the acyl groups were migrated to the 4-position at room temperature within an hour (Table 3,entry 2).The rear-rangement of p -acyloxy toluene also afforded regiospeci ?c prod-ucts,in which the acyl group was migrated to the 6-position.But the reaction time was longer than o -isomer in an identical manner described in Friedel e Crafts acylation (Table 3,entry 3).
The m -acyloxy toluene reactions afforded a more complex mixture than that of 4-and 6-isomers (Table 3,entry 4).Although the desired 4-and 6-orientated products (predominately 4-orien-tated product)were detected in the reaction mixture,the hydro-lysis product (m -cresol)was also detected when the reaction mixture was diluted in CDCl 3directly and then subjected to 1H NMR analysis.The maximum rearrangement was observed after 16h.
Our results indicated that the m -tolyl acetate 6was subjected to both Fries rearrangement and hydrolysis.The hydrolyzed m -cresol
Table 1
O-Acylation of phenols with acyl chlorides in 1%TfOH/CH 3
CN
R.Murashige et al./Tetrahedron 67(2011)641e 649
643
Table 2
Friedel e Crafts acylations of phenol derivatives with acyl chlorides in neat
TfOH
The reactions were set up equivalent of acyl halides at room temperature except parentheses.a
1.2equiv.b
1.5equiv.c
10equiv.d
At 60 C.e
At 100 C.
R.Murashige et al./Tetrahedron 67(2011)641e 649
644
reacted with an in situ generated acyl donor equivalent to a Frie-del e Crafts type reaction.To support the hypothesis,the reaction of m -cresol 7with stoichiometric amounts of acetic acid 8was com-pared with Fries rearrangement of 6in neat TfOH at room tem-perature (Scheme 4).Fries rearrangement of 6(82%)was faster than the reactions of 7and 8(52%)as monitored in an hour.But 16h later,the yields of both reactions became almost the same.The proportions of isomers (9and 10)did not change in any reaction time.The results indicated competitive reactions of Fries rear-rangement and hydrolysis of m -tolyl acetate 6were occurring,but the hydrolyzed acyl donor equivalent was also re-reacted with m -cresol as a Friedel e Crafts type reaction over a long reaction time.The results are consistent with the fact that stoichiometric amounts of acetic acid 8can act as an acyl donor for Friedel e Crafts acylation of toluene in TfOH.18However,a previous study reported that mainly o -acylated isomer was afforded by the treatment of m -cresol and carboxylic acid with graphite and methanesulfonic acid at 120 C in several hours.19On the other hand,our conditions,which were carried out at room temperature,afforded 4-and 6-acetylated isomers with a 1.4e 2:1ratio.The reaction condition can be applied to various carbon chains of m -acyloxy toluenes in good yield (Table 3,entry 4).
4-Fluoro-,4-chloro-,and 4-bromo-phenylacetate or phenyl-benzoate have to be heated at 60 C in neat TfOH to achieve Fries rearrangements in good yield (Table 3,entries 5e 7).Fries rear-rangement of 4-iodo phenylacetate or phenylbenzoate afforded a complex mixture as monitored after several hours and the yield of desired products was very low (Table 3,entry 9).4-Methoxyphenyl acetate has to be heated at 100 C to achieve Friedel e Crafts acyla-tions.Methoxy group was also deprotected during the reaction (Table 3,entry 10).
1-or 2-Naphthyl acetate also proceeded through a Fries rear-rangement,which consisted of a normal orientation.But 1-or 2-naphthyl benzoate did not afford the rearrangement product (Table 3,entries 10and 11).The results are consistent with large excess benzoyl chloride (10equiv)being needed in direct Frie-del e Crafts reactions for both naphthols.2.4.Synthesis of o -and p -homotyrosine
The comprehensive studies of reactivities of phenol derivatives described above were also applied to synthesis of optically pure homotyrosine from phenol 1without deprotection of the phenolic alcohol.Phenol,1and 3equiv of N -TFA e Asp(Cl)e OMe 1120afforded N -TFA e Asp e OMe b -phenyl ester 12,which is the O -acylated
product,in excellent yield when using diluted TfOH at room tem-perature for an hour.The phenyl ester of amino acids has normally been prepared with carbodiimides as a promoter,and conversions have not been quantitative.
Fries rearrangement of phenyl ester 12proceeded in neat TfOH at room temperature for 16h in good yield.The rearrangement afforded two C -acylated isomers,p -(13)and o -(14)aroylbenzene derivatives.The proportion was calculated as 1.4:1for 13and 14,respectively.
Friedel e Crafts reaction of phenol 1and N -TFA e Asp(Cl)e OMe 11also afforded two C -acylated isomers 13and 14using neat TfOH at room temperature for an hour.The proportion of the regioisomer was almost the same.The reaction time of the Friedel e Crafts re-action was much faster than that of the Fries rearrangement.These results are identical with our ?ndings described in Scheme 4that Fries rearrangement caused hydrolysis of phenyl ester and in situ generated acyl donor equivalent was subjected to a Friedel e Crafts type reaction.
The benzylic carbonyl of 13and 14was reduced to methylene with Pd/C under the H 2atmosphere (15,16),followed by depro-tection of the both protecting groups under the acidic conditions to afford optically pure (ee >99%)hTyr derivatives (17,18)in excellent yields.
Ruth has already reported the synthesis of o -hTyr from kyn-renine ?ve decades ago.21To the best of our knowledge,few papers have been subsequently reported for the synthesis of o -hTyr.It is ?rst time that the aspartic acid derivatives are in-troduced to phenol derivatives using the TfOH.TfOH has both catalytic activities for Friedel e Crafts and Fries rearrangements,and high solubility for the amino acid derivatives in good yield.The asymmetric center of the starting Asp derivatives in these products did not change (Scheme 5).
3.Conclusions
The effective synthetic routes for the O -acyloxy benzenes and hydroxyaryl ketones will be very useful for synthesis of bioactive compounds.Both skeletons were available from phenol derivatives and acyl equivalents in acidic conditions.TfOH is known as a super-acid and is used as catalyst in Friedel e Crafts acylation 22and al-kylation.23The acylation mechanisms proposed are that carboxylic acid derivatives form tri ?uoromethanesulfonic d carboxylic anhy-drides as active species,24and then the active species reacted with the aromatics.However few reports have been issued for the re-action with phenol derivatives.25The contribution of the TfOH for O-acylation of phenol derivatives,Friedel e Crafts acylation with acyl chloride,and Fries rearrangement of O -acyloxy benzenes was examined here.Our results show controls of O-acylation,Frie-del e Crafts acylation,and Fries rearrangement of acyloxy benzenes can be produced with TfOH proportion in CH 3CN.The O-acylation proceeded to completion at low concentration of TfOH in CH 3CN.On the other hand the Friedel e Crafts acylation and Fries rear-rangement almost proceeded effectively in neat TfOH.Further-more,our strategies are very bene ?cial because the reaction conditions were proceeded at milder conditions than previous reports.17,18,26
These observations on control,of these types of reactions were applied to hTyr synthesis,which is not easily prepared using pre-vious methods.Our synthetic routes produce good total yields and are easy for stereocontrolled synthesis,because optical purity of Asp derivatives can be maintained during the reaction.
The controls of O-acylation,Friedel e Crafts reactions,and Fries rearrangements of phenol derivatives by TfOH concentrations will contribute to effective preparation of the O -acyloxy benzenes and hydroxyaryl
ketones.
Scheme 3.Fries rearrangement of phenyl acetate (3)in various TfOH proportion in CH 3CN for an hour.
R.Murashige et al./Tetrahedron 67(2011)641e 649645
Table 3
Fries rearrangement of O -acyloxy benzenes and naphthalenes in neat
TfOH
The reactions were set up equivalent of acyl halides at room temperature except parentheses.a
At 60 C.b
At 100 C.c
At 100 C.d
At 100 C.e
At 100 C.
R.Murashige et al./Tetrahedron 67(2011)641e 649
646
4.Experimental section 4.1.General
NMR spectra were measured by JEOL ECA-500spectrometers.IR spectra were measured by Jasco FTIR-4100instrument.TfOH was purchased from Wako Chemicals.All solvents were of reagent grade and distilled using the appropriate methods.MS data were obtained with a Hitachi NanoFrontier LD mass spectrometer.Chiral HPLC was performed with Chirobiotic T (Astec), 4.6?250mm,eluted with 10%EtOH/H 2O;?ow rate,1.0ml/min;UV detection at 210nm.
4.2.General procedure for O-acylation of phenols in TfOH Phenol (0.28mmol)and acyl chloride (0.84mmol,3equiv)were dissolved in 1%TfOH/CH 3CN (1ml)at room temperature.The re-action mixture was stirred at same temperature for an hour,then poured into cold water and ethyl acetate.The organic layer was
washed with 1M HCl,saturated NaHCO 3,and saturated NaCl,and dried over MgSO 4,then ?ltered.The ?ltrate was concentrated to afford O -acylated products.All spectral data were identical with the literatures.27
4.3.General procedure of Friedel e Crafts acylation of phenols in TfOH
Phenol (0.28mmol)and acyl chloride (0.28mmol)were dis-solved in TfOH (3ml)at 0 C.The reaction mixture was warmed to room temperature for appropriate time in Table 2,then poured into cold water and ethyl acetate.The organic layer was washed with 1M HCl,saturated NaHCO 3,and saturated NaCl,and dried over MgSO 4,then ?ltrated.The ?ltrate was concentrated and the residue was subjected silica column chromatography to afford acylated products.All spectral data were identical with the literatures.274.4.General procedure of Fries rearrangement for O -acyloxy benzenes in TfOH
O -Acyloxy benzenes (0.28mmol)were dissolved in TfOH (3ml)at 0 C.The reaction mixture was warmed to room temperature for appropriate time in Table 3,then poured into cold water and ethyl acetate.The organic layer was washed with 1M HCl,saturated NaHCO 3,and saturated NaCl,and dried over MgSO 4,then ?ltrated.The ?ltrate was concentrated and the residue was subjected silica column chromatography to afford acylated products.All spectral data were identical with the literatures.264.5.Synthesis of optical pure homotyrosine
4.5.1.(S)-and (R)-1-Methyl 4-phenyl 2-(2,2,2-tri ?uoroacetamido)succinate (12).Phenol 1(30m l,0.34mmol)and (S )-11(0.267g,1.02mmol)were dissolved in 1ml of 1%TfOH in CH 3CN at 0 C.The reaction mixture was allowed to warm to room temperature and stirred for an hour,then poured into cold water and ethyl acetate.The organic layer was washed with saturated NaHCO 3,saturated NaCl,and dried over MgSO 4,then ?ltrated.The ?ltrate was con-centrated to afford (S )-12(0.098g,91%).
Compound (S )-12:d H (500MHz,CDCl 3)7.42e 7.39(3H,m),7.26(1H,t,J 7.4Hz),7.08(2H,d,J 8.0Hz),4.96(1H,m),3.84(3H,s,3H),3.38(1H,dd,J 17.8,4.0Hz),3.18(1H,dd,J 17.5,4.3Hz);d C (125MHz,CDCl 3)169.43,169.30,157.03(q,2J CF 38.0Hz),129.60,126.42,121.21,115.38(q,1J CF 300.3Hz),53.50,48.91,35.67;HRMS (ESI):MH t,found 320.0730.C 13H 13F 3NO 5requires 320.0746;[a ]D t86.0(c 1.0,CHCl 3).
Compound (R )-12was obtained from (R )-11in the same manner as (S )-12(86%).The d H and d c data for these samples were identical with these recorded for (S )-12;[a ]D à86.0(c 1.0,CHCl 3).
4.5.2.General procedure of Friedel e Crafts reaction for phenol 1and N-TFA e Asp(Cl)e OMe 11in TfOH.Phenol 1(0.02ml,0.23mmol)and (S )-11(60.3mg,0.23mmol)were dissolved in TfOH (1ml)at 0 C.The reaction mixture was warmed to room temperature,main-tained the temperature for an hour,and poured into cold water and ethyl acetate.The organic layer was washed with 1N HCl,5%NaHCO 3,1N HCl,and saturated NaCl successively,dried over MgSO 4,?ltrated,and concentrated.The residue was subjected to silica column chromatography (ethyl acetate/hexane ?1:2)to afford pure (S )-13and (S )-14as colorless amorphous mass.
Compound (S )-13:41%;n max (neat)3300,1714,1663,1600cm à1;d H (CDCl 3)7.85(2H,d,J 8.6Hz),7.58(1H,d,J 8.0Hz),6.89(2H,d,J 8.6Hz),4.97e 4.93(1H,m),3.83(1H,dd,J 18.3,4.0Hz),3.79(3H,s),3.51(1H,dd,J 18.3,4.0Hz);d C (CDCl 3)195.7,170.2,161.2,157.0(q,2
J CF ?38.0Hz),130.9,128.43,115.8(q,1J CF ?287.5Hz),115.6,
53.3,
Scheme 4.Time courses of Fries rearrangement of m -tolyl acetate (6)and Friedel e Crafts reaction with stoichiometric amounts of m -cresol (7)and acetic acid (8)in TfOH at room temperature.Yield was calculated as mixtures of 9and 10.
12
13, 15, 17 R 1=OH, R 2=H
14, 16, 18 R 1=H, R 2=OH
11
CH 331
231
2
1
2
+
i)
Scheme 5.Homotyrosine synthesis with Friedel e Crafts acylation and Fries re-arrangement.(i)3%TfOH,CH 3CN,room temperature,1h,86e 91%,(ii)TfOH,room temperature,1h,79%(13/14?1:1),(iii)TfOH,room temperature,16h,78e 84%(13/14?1.4:1),(iv)H 2,Pd/C,room temperature,1h,86e 99%,(v)6N HCl,80 C,6h,90e 97%.
R.Murashige et al./Tetrahedron 67(2011)641e 649647
48.7,39.3;HRMS(ESI):MHt,found320.0751.C13H13F3NO5requires 320.0746;[a]Dt75.0(c1.0,CHCl3).
Compound(S)-14:38%;n max(neat)3300,1720,1640, 1580cmà1;d H(500MHz,CDCl3)7.70(1H,d,J8.0Hz),7.53(1H,t,J 8.0Hz),7.44(1H,d,J6.9Hz),7.01(1H,d,J8.6Hz),6.95(1H,t, J?7.4Hz),4.96(1H,m),3.90(1H,dd,J18.3,4.0Hz),3.81(3H,s), 3.68(1H,dd,J18.3,4.0Hz);d C(125MHz,CDCl3)202.56,169.72, 162.45,156.98(q,2J CF38.0Hz),137.49,129.80,119.44,118.75, 118.59,115.50(q,1J CF287.5Hz),53.32,48.29,39.41;HRMS(ESI): MHt,found320.0749.C13H13F3NO5requires320.0746;[a]Dt107.0 (c1.0,CHCl3).
Compounds(R)-13and(R)-14were prepared in an identical manner as described above starting from(R)-11.The d H and d C data for these samples were identical with these recorded for(S)-13and (S)-14.
Compound(R)-13:40%;[a]Dà74.0(c1.0,CHCl3).
Compound(R)-14:39%;[a]Dà108.0(c1.0,CHCl3).
4.5.3.General procedure of Fries rearrangement in https://www.doczj.com/doc/49190867.html,pound (S)-12(1.008g,3.15mmol)was dissolved in TfOH(5ml)at0 C and warmed to room temperature.The reaction mixture was stirred for 16h and poured into cold water and ethyl acetate.The organic layer was treated in the same manner as described for Friedel e Crafts reaction to afford(S)-13(0.483g,48%)and(S)-14(0.360g,36%). Analytical data of each isomer were identical with the Friedel e Crafts products.
Compounds(R)-13and(R)-14were prepared in an identical manner as described above starting from(R)-12((R)-1345%,(R)-14 33%).Analytical data of each isomer were identical with the Frie-del e Crafts products.
4.5.4.General procedure for benzylic carbonyl https://www.doczj.com/doc/49190867.html,pound (S)-13(0.170g,0.53mmol)and Pd/C(10%,30mg)were suspended in acetic acid(10ml).The reaction mixture was stirred under hy-drogen atmosphere for an hour,and then?ltrated with Celite.The ?ltrate was concentrated and the residue was subjected to silica column chromatography(ethyl acetate/hexane?1:2)to afford(S)-15as amorphous mass(0.157g,97%).
Compound(S)-15:n max(neat)3350,1720,1595cmà1;d H (500MHz,CDCl3)7.03(2H,d,J8.6Hz),6.83(1H,d,J6.3Hz),6.77 (2H,d,J8.6Hz),4.67e4.64(1H,m),3.77(3H,s),2.61e2.58(2H,m), 2.27e2.24(1H,m),2.12e2.05(1H,m);d C(125MHz,CDCl3)171.40, 156.90(q,2J CF37.6Hz),154.29,131.58,129.41,115.53(q,1J CF 287.9Hz),115.45,52.96,52.37,33.33,30.39;HRMS(ESI):MHt, found306.0960.C13H15F3NO4requires306.0953;[a]Dt39.0(c1.0, CHCl3).
Compound(S)-16was obtained from(S)-14in the same manner as(S)-13.
Compound(S)-16:86%;n max(neat)3320,1720,1550cmà1;d H (500MHz,CDCl3)7.23(1H,d,J6.9Hz),7.10e7.09(2H,m),6.88(1H, t,J7.4Hz),6.74(1H,d,J8.0Hz),4.67e4.66(1H,m),3.68(3H,s), 2.72e2.67(2H,m), 2.35e2.26(1H,m), 2.19e2.16(1H,m);d C (125MHz,CDCl3)171.38,157.12(q,2J CF37.6Hz),153.52,130.52, 127.93,126.11,120.97,115.68(q,1J CF287.5Hz),115.45,52.87,52.37, 31.35,25.36;[a]Dt44.0(c1.0,CHCl3).
Compounds(R)-15and(R)-16were prepared in an identical manner as described above starting from corresponding pre-cursors.The d H and d C for these samples were identical with these recorded for(S)-15and(S)-16.
Compound(R)-15:99%;[a]Dà38.5(c1.0,CHCl3).
Compound(R)-16:87%;[a]Dà44.0(c1.0,CHCl3).
4.5.5.General procedure for https://www.doczj.com/doc/49190867.html,pound(S)-15 (0.152g,0.50mmol)was dissolved in6M HCl(2ml).The reaction mixture was heated at80 C for6h,then concentrated.The residue was subjected to silica column chromatography(CH3CN/MeOH/H2O?4:1:1)to afford(S)-17as colorless amorphous mass(0.103g, 90%).
Compound(S)-17:n max(neat)3100,1740cmà1;mp217e220 C;
d H(500MHz,CD3OD)7.06(2H,d,J8.6Hz),6.73(2H,d,J8.6Hz),
3.95(1H,t,J 6.3Hz), 2.75e2.63(2H,m), 2.22e2.18(1H,m), 2.13e2.06(1H,m);d C(125MHz,CD3OD)171.78,157.05,131.90, 130.36,116.41,53.44,33.92,31.23;HRMS(ESI):MHt,found 196.0958.C10H14NO3requires196.0974;[a]Dt35.6(c1.0,CHCl3); chiral HPLC t R?6.78min.
Compound(S)-18:95%;n max(neat)3350,3100,1750cmà1;mp 194e196 C;d H(500MHz,CD3OH)7.11(1H,d,J7.4Hz),7.06(1H,t,J 7.7Hz),6.80e6.77(2H,m),3.91(1H,t,J6.3Hz),2.85e2.82(1H,m), 2.75e2.69(1H,m),2.27e2.10(2H,m);d C(125MHz,CD3OD)172.82, 157.21,132.16,129.77,128.20,121.78,116.90,54.55,32.96,27.77;
[a]Dt27.3(c1.0,CHCl3);chiral HPLC t R?7.34min.
Compounds(R)-17and(R)-18:The d H and d C NMR for these samples were identical with these recorded for(S)-17and(S)-18.
Compound(R)-17:97%;[a]Dà34.5(c1.0,1N HCl);chiral HPLC t R?https://www.doczj.com/doc/49190867.html,pound(R)-18:96%;[a]Dà29.5(c1.0,1N HCl); chiral HPLC t R?9.35min.
Acknowledgements
We are very grateful to Professor G.D.Holman(University of Bath,U.K.)for valuable advice throughout the manuscript.This research was partially supported by Ministry of Education,Science, Sports and Culture Grant-in-Aid for Scienti?c Research on a Priority Area,18032007,for Scienti?c Research on Innovative Areas, 20200038and for Scienti?c Research(C),19510210,21510219.
Supplementary data
Supplementary data associated with this article can be found in online version at doi:10.1016/j.tet.2010.11.047.
References and notes
1.(a)Martin,https://www.doczj.com/doc/49190867.html,.Prep.Proced.Int.1992,24,369e435;(b)Olah,G.A.Frie-
del e Crafts and Related Reactions;Wiley-Interscience:New York,NY and London, 1963e1964;Vols.I e IV.
2.Olah,G.A.;Prahash,G.K.S.;Sommer,J.Superacids;Wiley:New York,NY,1985.
3.Olah,G.A.;Prahash,G.K.S.;Sommer,J.Science1979,206,13e20.
4.Kobayashi,S.;Sugiura,M.;Kitagawa,H.;Lam,W.W.L.Chem.Rev.2002,102,
2227e2302.
5.(a)Murashige,R.;Hayashi,Y.;Hashimoto,M.Tetrahedron Lett.2008,49,
6566e6568;(b)Murai,Y.;Hatanaka,Y.;Kanaoka,Y.;Hashimoto,M.Heterocy-cles2009,79,359e364;(c)Murashige,R.;Murai,Y.;Hatanaka,Y.;Hashimoto, M.Biosci.Biotechnol.Biochem.2009,73,1377e1380.
6.(a)Okumura,H.S.;Philmus,B.;Portmann,C.;Hemscheidt,T.K.J.Nat.Prod.
2009,72,172e176;(b)Plaza,A.;Bewley,https://www.doczj.com/doc/49190867.html,.Chem.2006,71,6898e6907.
7.Canesi,S.;Bouchu, D.;Ciufolini,M. A.Angew.Chem.,Int.Ed.2004,43,
4336e4338.
8.Wang,D.;Cole,P.A.J.Am.Chem.Soc.2001,123,8883e8886.
9.Evans,W.C.;Walker,N.J.Chem.Soc.1947,1571e1573.
10.Barfoot,C.W.;Harvey,J.E.;Kenworthy,M.N.;Kilburn,J.P.;Ahmed,M.;Taylor,
R.J.K.Tetrahedron2005,61,3403e3417.
11.Yamada,M.;Nagashima,N.;Hasegawa,J.;Takahashi,S.Tetrahedron Lett.1998,
39,9019e9022.
12.Xie,Y.;Lou,R.;Li,Z.;Mi, A.;Jiang,Y.Tetrahedron:Asymmetry2000,11,
1487e1494.
13.Brea,R.J.;L o pez-Deber,M.P.;Castedo,L.;Granja,https://www.doczj.com/doc/49190867.html,.Chem.2006,71,
7870e7873.
14.(a)Reifenrath,W.G.;Bertelli,D.J.;Micklus,M.J.;Fries,D.S.Tetrahedron Lett.
1976,17,1959e1962;(b)Nordlander,J.E.;Payne,M.J.;Njoroge,F.G.;Vish-wanath,V.M.;Han,G.R.;Laikos,G.D.;Balk,https://www.doczj.com/doc/49190867.html,.Chem.1985,50, 3619e3622;(c)Melillo,D.G.;Larsen,R.D.;Mathre,D.J.;Shukis,W.F.;Wood,
A.W.;Colleluori,https://www.doczj.com/doc/49190867.html,.Chem.1987,52,5143e5150;(d)Griesbeck,A.G.;
Heckroth,H.Synlett1997,1243e1244;(e)Lin,W.;He,Z.;Zhang,H.;Zhang,X.;
Mi,A.;Jiang,Y.Synthesis2001,1007e1009;(f)Xu,Q.;Wang,G.;Wang,X.;Wu, T.;Pan,X.;Chan,A.S.C.;Yang,T.Tetrahedron:Asymmetry2000,11,2309e2314.
15.Dumeunier,R.;Mark o,I.E.Tetrahedron Lett.2004,45,825e829.
16.Kobayashi,S.;Moriwaki,M.;Hachiya,I.Tetrahedron Lett.1996,37,2053e2056.
17.(a)Kaboudin,B.Tetrahedron1999,55,12865e12872;(b)Mouhtady,O.;Gas-
pard-Iloughmane,H.;Roques,N.;Rouxa, C.L.Tetrahedron Lett.2003,44, 6379e6382.
R.Murashige et al./Tetrahedron67(2011)641e649 648
18.Roberts,R.M.G.;Sadri,A.R.Tetrahedron1983,39,137e142.
19.Sharghi,H.;Hosseini-Sarvari,M.;Eskandari,R.Synthesis2006,2047e2052.
20.Weygand,F.;Klinke,P.;Eigen,I.Chem.Ber.1957,90,1896e1905.
21.Ruth,W.Acta Chem.Scand.1954,8,1542e1546.
22.Effenberger, F.;Eberhard,J.K.;Maier, A.H.J.Am.Chem.Soc.1996,118,
12572e12579.
23.Booth,B.L.;Haszeldine,R.N.;Laali,K.J.Chem.Soc.,Perkin Trans.11980,
2887e2893.
24.(a)Effenberger,F.;Epple,G.Angew.Chem.,Int.Ed.Engl.1972,11,299e300;(b)
Effenberger,F.;Epple,G.Angew.Chem.,Int.Ed.Engl.1972,11,300e301.
25.Krawczyk,H.;Albrecht,L.;Wojciechowski,J.;Wolf,W.M.Tetrahedron2007,63,
12583e12594.26.Matsushita,Y.;Sugamoto,K.;Matsui,T.Tetrahedron Lett.2004,45,4723e4727.
27.(a)Lota,R.K.;Olusanjo,M.S.;Dhanani,S.;Owen,C.P.;Ahmed,S.J.Steroid
Biochem.Mol.Biol.2008,111,128e137;(b)Park,K.K.;Jeong,J.Tetrahedron2005, 61,545e553;(c)Armstrong,E.C.;Bent,R.L.;Loria,A.;Thirtle,J.R.;Weiss-berger,A.J.Am.Chem.Soc.1960,82,1928e1935;(d)Fischer,A.;Henderson,G.
N.;Thompson,R.J.Aust.J.Chem.1978,31,1241e1247;(e)Gu,W.;Hill,A.J.;
Wang,X.;Cui,C.;Weiss,R.G.Macromolecules2000,33,7801e7811;(f)Montiel-Smith,S.;Meza-Reyes,S.;Vi~n as-Bravo,O.;Fern a ndez-Herrera,M.A.;Martínez-Pascual,R.;Sandoval-Ramírez,J.;Fuente,A.;Reyes,M.;Ruiz,J.A.ARKIVOC2005, 127e135;(g)Liu,Z.;Sun,Y.;Wang,J.;Zhu,H.;Zhou,H.;Hu,J.;Wang,J.Sep.
Purif.Technol.2008,64,247e252;(h)Park,K.K.;Lee,H.J.;Kim,E.H.;Kang,S.K.
J.Photochem.Photobiol.,A:Chem.2003,159,17e21.
R.Murashige et al./Tetrahedron67(2011)641e649649
中国三氟甲磺酸行业市场前景调查及投融资战略研究报告 2017-2022年
前言 企业成功的关键就在于,能否跳出红海,开辟蓝海。那些成功的公司往往都会倾尽毕生的精力及资源搜寻产业的当前需求、潜在需求以及新的需求!随着行业竞争的不断加剧,大型企业间并购整合与资本运作日趋频繁,国内外优秀的企业愈来愈重视对行业市场的研究,特别是对企业发展环境和客户需求趋势变化的深入研究,逐渐成为行业中的翘楚! 中商产业研究院发布《2017-2022年中国三氟甲磺酸行业市场前景调查及投融资战略研究报告》利用中商数据库长期对三氟甲磺酸行业市场跟踪搜集的相关数据,全面准确地为您从行业的整体高度架构分析体系。报告从行业的宏观环境出发,以三氟甲磺酸行业的产销状况和行业的需求走向为依托,详尽分析了近年中国三氟甲磺酸行业当前的市场容量、产销规模、发展速度和竞争态势;报告还同时分析了三氟甲磺酸行业进出口市场、行业的上下游产业链运营情况,行业市场需求特征等,并且对三氟甲磺酸行业市场领先企业经营状况进行分析,最后对未来几年三氟甲磺酸行业发展趋势与投资前景做出预测。
【出版日期】2016年 【交付方式】Email电子版/特快专递 【价格】纸介版:25800元电子版:25500元纸介+电子:25800元 第一章三氟甲磺酸产业概述 一、三氟甲磺酸定义 二、三氟甲磺酸分类 三、三氟甲磺酸用途 四、三氟甲磺酸经营模式 第二章全球及中国三氟甲磺酸市场分析 第一节三氟甲磺酸行业国际市场分析 一、三氟甲磺酸重点生产企业 二、三氟甲磺酸产品技术动态 三、三氟甲磺酸竞争格局分析 四、三氟甲磺酸国际市场前景 第二节三氟甲磺酸行业国内市场分析 一、三氟甲磺酸国内市场现状 二、三氟甲磺酸产品技术动态 三、三氟甲磺酸竞争格局分析 四、三氟甲磺酸国内需求现状 五、三氟甲磺酸国内市场趋势 第三节三氟甲磺酸国内外市场对比分析 第三章三氟甲磺酸行业市场环境分析 一、国际宏观经济及前景预测 (一)国际宏观经济环境分析 (二)国际经济市场前景分析 二、国内宏观经济及前景预测 (一)中国宏观经济环境分析 (二)中国经济市场前景展望 第四章三氟甲磺酸行业相关政策分析 一、三氟甲磺酸行业监管体制 二、三氟甲磺酸行业政策分析 三、三氟甲磺酸相关标准分析
三氟甲基磺酸研发技术报告 1 三氟甲基磺酸性能简介 三氟甲基磺酸又名三弗里克酸,英文名称:trifluoromethanesulfonic acid,分子式:CF3SO3H,CAS号:1493-13-6,m.p:-40℃,b.p:162℃,密度:1.696g/ml(25℃),折色率:1.326(25℃),常温下具有强烈刺激性的无色透明液体。与水可以任意比例互溶,也溶于二甲基甲酰胺、环丁砜、二甲基亚砜以及丙烯腈等极性有机溶剂,在潮湿空气中生成稳定的一水合物。 2 三氟甲基磺酸的应用 三氟甲基磺酸与其他超强酸不同,有着极高的耐热性、耐氧化-还原性。此外,在强亲核剂的存在下,也不会游离出氟离子,故有无卤液体有机强酸的功能。并且,其共轭碱三氟磺酸离子(CF3SO3-)因负电荷分布于构成离子的所有原子,故有特殊的非亲核性、非配位性的特征。因此,三氟甲基磺酸在一系列化学过程中用作催化剂或反应物有很大的优点。 2.1 作酸催化剂 三氟甲基磺酸可在多种反应中用作催化剂。在弗里德尔-克拉茨反应中,用作烷基化和酰基化的催化剂,也能使无活性的芳族化合物进行催化,与其他催化剂相比,能以较少用量的催化剂高收率的产生酰基化合物。对于Gattermann-Koch 型甲基化反应、羧酸合成、亚氨离子反应等许多都有着优良的酸催化剂功能。此外,在酸催化重排中,可用作贝克曼重排、弗里斯重排、烷基和硝基重排等反应的催化剂。在苯乙烯烃聚合、硅氧烷衍生物聚合中也可用做酸性聚合催化剂,其效果显著,能提高反应速率和收率。三氟甲基磺酸除单独用作酸催化剂外,还能吸附于沸石、二氧化硅用作固体催化剂。 2.2 三氟甲基磺酸衍生物的应用 三氟甲基磺酸与金属氢氧化物或三氟甲基磺酸盐在水溶液发生放热反应,生成相应的金属盐,这些金属盐可溶于多种有机溶剂,对热极为稳定,如三氟甲基磺酸锂(CF3SO3Li)因热稳定性优良,以广泛用作电池的电解质。新型锂盐LiTFSI[(CF3SO2)2NLi]与传统锂盐LiFP6相比,具有更高的稳定性,其在新型电池的应用已得到国内外广泛关注。而以三氟阴离子为抗衡离子的锡化合物是醛醇
一种实用的、价廉的、环保的方法制备三氟甲磺酸铋 摘要:通过冷冻干燥,以三氧化二铋为原料,大规模制备三氟甲磺酸铋的水合物。 铋的衍生物吸引了不少有机化学家的注意。在过去的几年里,三氟甲磺酸铋曾被报道,可以作为一种新型和有效的有机催化剂。与其他已知的三氟甲磺酸金属化合物特别是与那些过渡金属元素形成的化合物相比较,三氟甲磺酸铋表现出很强的活性。但是,以前报道的制备这种化合物的过程有很大的缺陷,而且很难转为大规模:需要过量的昂贵的三氟甲磺酸试剂或者需要苛刻的反应条件。一条具有吸引力的路线就是只需要化学计量的三氟甲磺酸与三苯基铋的氢去金属化反应。然而,它需要使用一种完全商业化的溶剂和相当贵的三苯基铋,副产物是具有致癌性的苯。大部分的三氟甲磺酸金属,特别是稀土元素,都是在沸水中通过三氟甲磺酸直接与相应的金属氧化物反应制备。这种方法,也包括采用便宜的三氧化二铋作为起始原料,只要采用一点点三氟甲磺酸,就可以很有效的制备三氟甲磺酸铋。本文就报道了这种方法的结果。 首先,我们按照文献报道的制备三氟甲磺酸稀土元素的方法去做,以三氧化二铋为原料。不幸的是,这个过程中形成了水溶性的铋盐和白色的水不溶固体,即使采用非常纯的原料,也会出现这样的情况。通过X衍射分析发现,是形成了氧化铋的硫酸氢盐,产率大约是20%。不经意之间,我们发现化合物2,暴露在空气中,能很快转变为氧的碳酸铋盐。由于化合物1在一些有机溶剂中溶解,我们决定试着把这些溶剂与水混合。在经过多次试验后,我们发现乙醇和水75:25的条件下,能完全避免化合物2的形成,且能以较高的产率得到化合物1。令人惊讶的是,当反应在纯乙醇中进行时,反应不发生。减压蒸去溶剂,得到白色糊状物质,能溶于水。因为我们之前的研究表明化合物1是一种对温度很敏感的化合物,通过冷冻干燥法除去水分。在这些条件下,得到白色粉末。它经过红外、核磁的鉴定,所得数据与以前报道的相似。将冷冻干产物暴露于空气中能得到化合物1的九水合物,随后,加热到45度,会形成纯的五水合物。最后,用两种方法制备的三氟甲磺酸铋来催化甲苯的苯甲酰化,得到了相似的产物。 众所周知,铋盐在水中容易水解,产生多核阳离子。 铋与这些阳离子缩合形成的物质的pk h主要取决于相应的阴离子,浓度以及溶液PH。其他阳离子形式,如[Bi6O n(OH)12-2n]6+,是从高氯酸铋和硝酸铋中鉴定出来的。当化合物1
三氟甲磺酸金属盐催化构建碳-碳键反应的研究近年来,路易斯酸作为催化剂参与的有机合成反应引起了广泛的关注。但是,一些传统的路易斯酸仍存在一些缺点,如催化活性低;反应选择性差;对水敏感, 稳定性差易失活等。 因此,发展高效、高选择性和高稳定性的新型路易斯酸是现代有机合成发展的必然趋势。作为一种新型的路易斯酸,三氟甲磺酸金属盐具有强的热力学和化学稳定性以及路易斯酸性。 由于这些独特的性质,三氟甲磺酸金属盐已经被广泛地应用于一系列有机合成反应中。同时,碳-碳键的构建是有机合成方法学中最重要的研究内容之一。 在此基础上,本论文主要研究了三氟甲磺酸金属盐在构建碳-碳键反应中的 应用,主要涉及了多组分反应和烯丙基化反应。首先研究了三氟甲磺酸钪催化的多组分反应。 2-甲酰基苯甲酸甲酯、胺和三甲基氰硅烷可以在三氟甲磺酸钪催化下发生Strecker/分子内酰胺化串联反应,通过“一锅法”合成1-氰基取代的异吲哚啉 酮类衍生物。该反应只需添加2.5 mol%的催化剂,得到的目标产物收率最高达97%。 这种方法具有原料简单易得、实验操作简单、底物适用性好等优点。另外,该反应过程中不仅构建了新的碳-碳键,也构建了新的碳-氮键。 其次,探索了三氟甲磺酸铟催化的多组分反应。在三氟甲磺酸铟的催化下, 邻羧酸苯甲醛、胺和二氟烯醇硅醚发生Mannich/分子内酰胺化串联反应,构建新的碳-碳键和碳-氮键,生成收率最高达86%的1-二氟亚甲基取代异吲哚啉酮类衍生物。 此外,对甲氧基苯胺为原料合成的异吲哚啉酮可以在硝酸铈铵的作用下氧化
脱去对甲氧基苯基,生成具有仲胺结构的异吲哚啉酮。仲胺上NH的进一步转化为拓展1-二氟亚甲基取代异吲哚啉酮的分子多样性提供了条件。 第三,研究了三氟甲磺酸铜催化的氧化烯丙基化反应,制备了高烯丙基胺类 化合物。以氧气或叔丁基过氧化氢为氧化剂,甘氨酸衍生物在三氟甲磺酸铜的催化下可以发生α位的氧化烯丙基化反应。 在该过程中,甘氨酸衍生物首先脱去α位碳原子上的氢,氧化生成亚胺类中 间体。之后亚胺中间体在路易斯酸的活化下发生烯丙基化反应,形成新的碳-碳键。 甘氨酸酯和甘氨酸酰胺类物质都适用于该反应,能以41%-73%的产率生成α -烯丙基甘氨酸衍生物。控制实验证明,三氟甲磺酸铜不仅作为路易斯酸参与亚胺中间体的活化,也作为氧化剂参与了甘氨酸酯衍生物的氧化脱氢过程。 最后,研究了三氟甲磺酸铟催化的不对称烯丙基化反应。三氟甲磺酸铟可以与手性吡啶咪唑啉配体共同催化靛红亚胺的不对称烯丙基化反应,构建高立体选择性的碳-碳键。 该反应只需2.5 mol%的催化剂和配体,无需其它添加剂,在室温下反应得到 较高产率和ee值(最高97%)的3-烯丙基-3-氨基-2-吲哚酮类衍生物。此外,通过单晶结构的分析确定了产物的主要构型为S型。
三氟甲磺酸锂的制备及应用研究进展 摘要:综述了三氟甲磺酸锂的制法、市场及应用。由于锂离子电池的应用正迅速扩展到一切使用电池的地方,因而三氟甲磺酸锂也必将有良好的发展前景。 关键词:三氟甲磺酸锂制备市场应用 近几年,伴随着锂离子电池的快速发展,锂离子电池所需电解液的需求量也在迅速增加。为了满足锂离子电池产业未来发展的需要,必须开发出高安全性、高环境适应性的动力电池电解液材料。虽然目前liPF6(六氟磷酸锂盐)被公认为是较为理想的锂离子电池电解液,但LiPF6合成工艺复杂,分解温度低,从60 ℃开始就有少量分解,在较高温度或恶劣的环境下,分解的比例大大增加,产生HF(氢氟酸)等游离酸,从而使电解液酸化,最终导致电极材料的损坏以及电池性能的急剧恶化。CF3SO3Li(三氟甲磺酸锂)在热稳定性、吸水分解性、循环性能等方面都高于LiPF6,尤其是CF3SO3li应用于固体电解质时,由于其稳定的阴离子会使电解质和阴极材料界面间的钝化层结构和组成得到改善,有利于电解质、钝化膜和电机的稳定。因此,CF3SO3Li的生产和应用必将成为研究的热点。 1.三氟甲磺酸锂的性质与质量指标 1.1 物理性质 三氟甲基磺酸锂,又名三氟甲烷磺酸锂,分子式:CF3SO3Li;密度: 1.9 g/cm3;熔点:423 ℃;白色粉末,易吸潮,溶于水及其它部分溶剂时放热。 1.2 质量指标 3.市场及应用 3.1 市场情况 锂离子电池具有工作电压高、体积小、无记忆效应、无污染、自放电小、循环寿命长等优点,目前已广泛应用于笔记本电脑、手机、PDA、数码相机和携带式电动工具等领域。另外,受益于电动汽车推广,三氟甲磺酸锂有望呈现爆发式增长。 3.2 应用研究 目前,CF3SO3li的工业应用主要是以锂电池电解液为主。此外,固体聚合物电解质具有良好的柔韧性、成膜性、稳定性和成本低等特点,既可作为正负电极间隔膜用又可作为传递离子的电解质用,是CF3SO3li应用的又一重要研究领
推荐三氟甲磺酸项目可行性研究报告(技术工艺+设备选型+财务概 算+厂区规划)标准方案设计 【编制机构】:博思远略咨询公司(360投资情报研究中心) 【研究思路】:
【关键词识别】:1、三氟甲磺酸项目可研 2、三氟甲磺酸市场前景分析预测 3、三氟甲磺酸项目技术方案设计 4、三氟甲磺酸项目设备方案配置 5、三氟甲磺酸项目财务方案分析 6、三氟甲磺酸项目环保节能方案设计 7、三氟甲磺酸项目厂区平面图设计 8、三氟甲磺酸项目融资方案设计 9、三氟甲磺酸项目盈利能力测算 10、项目立项可行性研究报告 11、银行贷款用可研报告 12、甲级资质13、三氟甲磺酸项目投资决策分析 【应用领域】: 【三氟甲磺酸项目可研报告详细大纲——2013年发改委标准】: 第一章三氟甲磺酸项目总论 项目基本情况 项目承办单位 可行性研究报告编制依据 项目建设内容与规模 项目总投资及资金来源 经济及社会效益 结论与建议 第二章三氟甲磺酸项目建设背景及必要性
项目建设背景 项目建设的必要性 第三章三氟甲磺酸项目承办单位概况 公司介绍 公司项目承办优势 第四章三氟甲磺酸项目产品市场分析 市场前景与发展趋势 市场容量分析 市场竞争格局 价格现状及预测 市场主要原材料供应 营销策略 第五章三氟甲磺酸项目技术工艺方案 项目产品、规格及生产规模 项目技术工艺及来源 项目主要技术及其来源 项目工艺流程图 项目设备选型 项目无形资产投入 第六章三氟甲磺酸项目原材料及燃料动力供应主要原料材料供应 燃料及动力供应 主要原材料、燃料及动力价格 项目物料平衡及年消耗定额 第七章三氟甲磺酸项目地址选择与土建工程项目地址现状及建设条件 项目总平面布置与场内外运 总平面布置 场内外运输 辅助工程 给排水工程 供电工程 采暖与供热工程