
三种分析蛋白结构域(Domains)的方法
三种分析蛋白结构域(Domains)的方法
1,SMART入门,蛋白结构和功能分析
SMART介绍
SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART.
SMART(http://smart.embl-heidelberg.de/),可以说是蛋白结构预测和功能分析的工具集合。简单点说,就是集合了一些工具,可以预测蛋白的一些二级结构。如跨膜区(Transmembrane segments),复合螺旋区(coiled coil regions),信号肽(Signal peptides),蛋白结构域(PFAM domains)等。
SMART前该知道的
1,SMART有两种不同的模式:normal 或genomic
主要是用的数据库不一样。Normal SMART, 用的数据库 Swiss-Prot,
SP-TrEMBL 和 stable Ensembl proteomes。Genomic SMART, 用全基因组序列。详细列表:http://smart.embl-heidelberg.de/smart/list_genomes.pl
2,一些名词解释
http://smart.embl-heidelberg.de/help/smart_glossary.shtml
SMART进行时
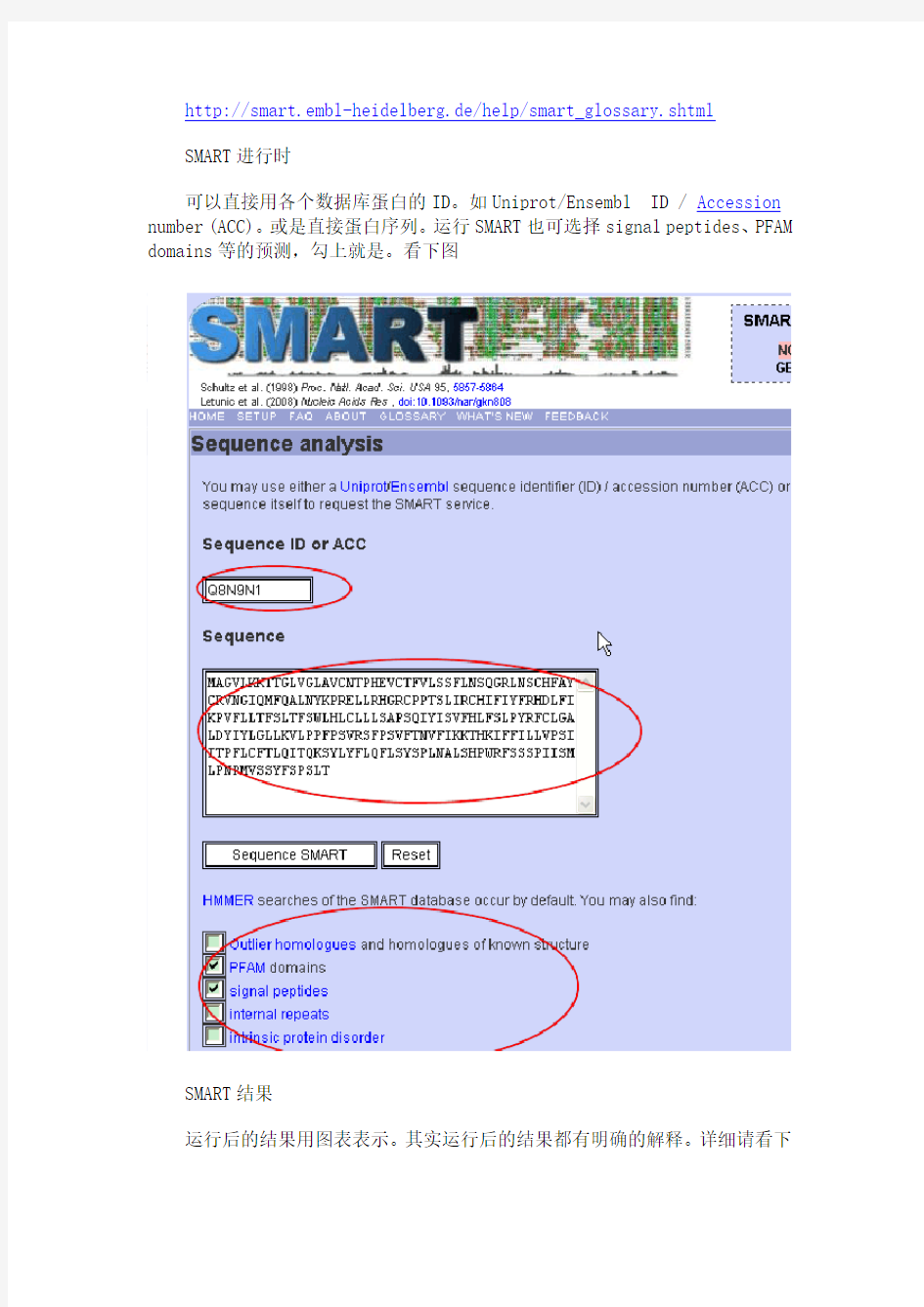
可以直接用各个数据库蛋白的ID。如Uniprot/Ensembl ID / Accession number (ACC)。或是直接蛋白序列。运行SMART也可选择signal peptides、PFAM domains等的预测,勾上就是。看下图
SMART结果
运行后的结果用图表表示。其实运行后的结果都有明确的解释。详细请看下
面。
不同结构的预测由不同的工具完成。如果你想了解更多,可访问去该工具的网站。
?跨膜区(Transmembrane segments), TMHMM2 program 。(用表示 ) ?复合螺旋区(coiled coil regions),Coils2 program。( 用表示) ?信号肽(Signal peptides),SignalP program。( )
?蛋白结构域(PFAM),PFAM。
等等。。不止这几个的。其它不一一列举。因为都是详细的说明。点击图标链接,就能看到该区域的序列,或是一些详细的描述。如上图的跨膜区,点击进去就是该跨膜区从开始到结束的序列。
另外,不一定所有预测的区域都会用在图示里看到。一般SMART的显示顺序是SMART > PFAM > PROSPERO repeats > Signal peptide > Transmembrane > Coiled coil > Unstructured regions > Low complexity。另外其它不用图解显示的区域,在底下的表格也有详细说明。
2,Sanger的Pfam数据库
网址:https://www.doczj.com/doc/378548622.html,/
目前的版本:Pfam 23.0 (July 2008, 10340 families)
The Pfam database is a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs).
3,NCBI的CDD(Conserved Domain Database)数据库
网址:https://www.doczj.com/doc/378548622.html,/Structure/cdd/wrpsb.cgi
Proteins often contain several modules or domains, each with a distinct evolutionary origin and function. NCBI’s Conserved Domain Database is a collection of multiple sequence alignments for ancient domains and full-length proteins.
最后,自己试验一下。上面两个图的结果的数据是用了NP_776850的蛋白序列。你也可以拿这个序列来运行一下看看。
三种分析蛋白结构域(Domains)的方法 1,SMART入门,蛋白结构和功能分析 SMART介绍 SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART. SMART(,可以说是蛋白结构预测和功能分析的工具集合。简单点说,就是 集合了一些工具,可以预测蛋白的一些二级结构。如跨膜区(Transmembrane segments),复合螺旋区(coiled coil regions),信号肽(Signal peptides),蛋白结构域(PFAM domains)等。 SMART前该知道的 1,SMART有两种不同的模式:normal 或genomic 主要是用的数据库不一样。Normal SMART, 用的数据库 Swiss-Prot, SP-TrEMBL 和 stable Ensembl proteomes。Genomic SMART, 用全基因组序列。详细列表:,一些名词解释 进行时 可以直接用各个数据库蛋白的ID。如Uniprot/Ensembl??ID / Accession number (ACC)。或是直接蛋白序列。运行SMART也可选择signal peptides、PFAM domains等的预测,勾上就是。看下图 SMART结果 运行后的结果用图表表示。其实运行后的结果都有明确的解释。详细请看下面。
大脑结构与功能 大脑结构详解
大脑(Brain)包括左、右两个半球及连接两个半球的中间部分,即第三脑室前端的终板。大脑半球被覆灰质,称大脑皮质,其深方为白质,称为髓质。髓质内的灰质核团为基底神经节。在大脑两半球间由巨束纤维—相连。 具体内容有大脑半球各脑叶、大脑皮质功能定位、大脑半球深部结构、大脑半球内白质、嗅脑和边缘系统五大部分。 各叶的位臵、结构和主要功能如下: 1、额叶:也叫前额叶。位于中央沟以前。在中央沟和中央前沟之间为中央前回。在其前方有额上沟和饿下沟,被两沟相间的是额上回、额中回和额下回。额下回的后部有外侧裂的升支和水平分支分为眶部、三角部和盖部。额叶前端为额极。额叶底面有眶沟界出的直回和眶回,其最内方的深沟为嗅束沟,容纳嗅束和嗅球。嗅束向后分为内侧和外侧嗅纹,其分叉界出的三角区称为嗅三角,也称为前穿质,前部脑底动脉环的许多穿支血管由此入脑。在额叶的内侧面,中央前、后回延续的部分,称为旁中央小叶。负责思维、计划,与个体的需求和情感相关。 2、顶叶:位于中央沟之后,顶枕裂于枕前切迹连线之前。在中央沟和中央后沟之间为中央后回。横行的顶间沟将顶叶余部分为顶上小叶和顶下小叶。顶下小叶又包括缘上回和角回。响应疼痛、触摸、品尝、温度、压力的感觉,该区域也与数学和逻辑相关。 3、颞叶:位于外侧裂下方,由颞上、中、下三条沟分为颞上回、颞中回、颞下回。隐在外侧裂内的是颞横回。在颞叶的侧面和底面,在颞下沟和侧副裂间为梭状回,,侧副裂与海马裂之间为海马回,围绕海马裂前端的钩状部分称为海马钩回。负责处理听觉信息,也与记忆和情感有关。 4、枕叶位于枕顶裂和枕前切迹连线之后。在内侧面,,距状裂和顶枕裂之间为楔叶,与侧副裂候补之间为舌回。负责处理视觉信息。 5、岛叶:位于外侧裂的深方,其表面的斜行中央钩分为长回和短回。 6、边缘系统:与记忆有关,在行为方面与情感有关。 大脑的总结构 大脑皮质为中枢神经系统的最高级中枢,各皮质的功能复杂,不仅与躯体的各种感觉和运动有关,也与语言、文字等密切相关。根据大脑皮质的细胞成分、排列、构筑等特点,将皮质分为若干区。 现在按Brodmann提出的机能区定位简述如下: ·皮质运动区:位于中央前回(4区),是支配对侧躯体随意运动的中枢。它主要接受来自对侧骨骼肌、肌腱和关节的本体感觉冲动,以感受身体的位臵、姿势和运动感觉,并发出纤维,即锥体束控制对侧骨骼肌的随意运动。返回皮质运动前区:位于中央前回之前(6区),为锥体外系皮质区。它发出纤维至丘脑、基底神经节、红核、黑质等。与联合运动和姿势动作协调有关,也具有植物神经皮质中枢的部分功能。 ·皮质眼球运动区:位于额叶的8枢和枕叶19区,为眼球运动同向凝视中枢,管理两眼球同时向对侧注视。皮质一般感觉区:位于中央后回(1、2、3区),接受身体对侧的痛、温、触和本体感觉冲动,并形成相应的感觉。顶上小叶(5、
结构域 科技名词定义 中文名称:结构域 英文名称:domain;structural domain;motif 其他名称:模体,基序 定义1:多肽链内一段类似球形的折叠区。多数结构域具有一定的一级结构和相应功能。 所属学科:免疫学(一级学科);概论(二级学科);免疫学相关名词(三级学科) 定义2:蛋白质或核酸分子中含有的、与特定功能相关的一些连续的或不连续的氨基酸或核苷酸残基。 所属学科:生物化学与分子生物学(一级学科);总论(二级学科) 定义3:蛋白质多肽链中可被特定分子识别和具有特定功能的三级结构元件。 所属学科:细胞生物学(一级学科);细胞化学(二级学科) 本内容由全国科学技术名词审定委员会审定公布 结构域是生物大分子中具有特异结构和独立功能的区域,特别指蛋白质中这样的区域。在球形蛋白中,结构域具有自己特定的四级结构,其功能部依赖于蛋白质分子中的其余部分,但是同一种蛋白质中不同结构域间常可通过不具二级结构的短序列连接起来。蛋白质分子中不同的结构域常由基因的不同外显子所编码。 目录 编辑本段介绍 (Domain)
在蛋白质三级结构内的独立折叠单元。结构域通常都是几个超二级结构单元的组合 结构域 。 结构域(Structural Domain)是介于二级和三级结构之间的另一种结构层次。所谓结构域是指蛋白质亚基结构中明显分开的紧密球状结构区域,又称为辖区。多肽链首先是在某些区域相邻的氨基酸残基形成有规则的二级结构,然后,又由相邻的二级结构片段集装在一起形成超二级结构,在此基础上多肽链折叠成近似于球状的三级结构。对于较大的蛋白质分子或亚基,多肽链往往由两个或多个在空间上可明显区分的、相对独立的区域性结构缔合而成三级结构,这种相对独立的区域性结构就称为结构域。对于较小的蛋白质分子或亚基来说,结构域和它的三级结构往往是一个意思,也就是说这些蛋白质或亚基是单结构域。结构域自身是紧密装配的,但结构域与结构域之间关系松懈。结构域与结构域之间常常有一段长短不等的肽链相连,形成所谓铰链区。不同蛋白质分子中结构域的数目不同,同一蛋白质分子中的几个结构域彼此相似或很不相同。常见结构域的氨基酸残基数在100~400个之间,最小的结构域只有40~50个氨基酸残基,大的结构域可超过400个氨基酸残基。 编辑本段连接状况 有些球 结构域 形蛋白的一条肽链,或以共价键相连的两条或多条肽链在空间结构上可以区分为若干个球状的子结构,其中的每一个球状子结构就被称为一个结构域。
蛋白质结构解析的方法对比综述 工程硕士李瑾 摘要:到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法,这两种方法各有优点和不足。 关键词:x射线衍射法 NMR法 到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法。其中X射线的方法产生的更早,也更加的成熟,解析的数量也更多,第一个解析的蛋白的结构,就是用x晶体衍射的方法解析的。而NMR方法则是在90年代才成熟并发展起来的。这两种方法各有优点和不足[1]。 首先是X射线晶体衍射法。该方法的前提是要得到蛋白质的晶体。通常是将表达目的蛋白的基因经PCR扩增后克隆到一种表达载体中,然后转入大肠杆菌中诱导表达,目的蛋白提纯之后摸索结晶条件,等拿到晶体之后,将晶体进行x射线衍射,收集衍射图谱,通过一系列的计算,得到蛋白质的原子结构[2]。 x射线晶体衍射法的优点是:速度快,通常只要拿到晶体,最快当天就能得出结构,另外不受肽链大小限制,无论是多大分子量的蛋白质或者RNA、DNA,甚至是结合多种小分子的复合体,只要能够结晶就能够得到其原子结构。所以x射线方法解析蛋白的关键是摸索蛋白结晶的条件。该方法得到的是蛋白质分子在晶体状态下的空间结构,这种结构与蛋白质分子在生物细胞内的本来结构有较大的差别。晶体中的蛋白质分子相互间是有规律地、紧密地排列在一起的,运动性较差;而自然界的生物细胞中的蛋白质分子则是处于一种溶液状态,周围是水分子和其他的生物分子,具有很好的运动性。而且,有些蛋白质只能稳定地存在于溶液状态,无法结晶[2]。 核磁共振NMR(nuclear magnetic resonance)现象很早就被科研人员观察到了,但将这种方法用来解析蛋白质结构,却是近一二十年的事情。NMR法具体原理是对水溶液中的蛋白质样品测定一系列不同的二维核磁共振图谱,然后根据已确定的蛋白质分子的一级结构,通过对各种二维核磁共振图谱的比较和解析,在图谱上找到各个序列号氨基酸上的各种氢原子所对应的峰。有了这些被指认的峰,就可以根据这些峰在核磁共振谱图上所呈现的相互之间的关系得到它们所对应的氢原子之间的距离。[3]可以想象,正是因为蛋白质分子具有空间结构,在序列上相差甚远的两个氨基酸有可能在空间距离上是很近的,它们所含的氢原子所对应的NMR峰之间就会有相关信号出现[4] 。通常,如果两个氢原子之间距离小于0.5纳米的话,它们之间就会有相关信号出现。一个由几十个氨基酸残基组成的蛋白质分子可以得到几百个甚至几千个这样与距离有关的信号,按照信号的强弱把它们转换成对应的氢原子之间的距离,然后运用计算机程序根据所得到的距离条件模拟出该蛋白质分子的空间结构。该结构既要满足从核磁共振图谱上得到的所有距离条件,还要满足化学上有关原子与原子结合的一些基本限制条件,如原子间的化学键长、键角和原子半径等[4]。 NMR解析蛋白结构常规步骤如下:首先通过基因工程的方法,得到提纯的目的蛋白,在蛋白质稳定的条件下,将未聚合,而且折叠良好的蛋白样品(通常是1mM-3mM,500ul,PH6-7的PBS)装入核磁管中,放入核磁谱仪中,然后由写好的程序控制谱仪,发出一系列的电磁波,激发蛋白中的H、13N、13C原子,等电磁波发射完毕,再收集受激发的原子所放出的“能量”,通过收集数据、谱图处理、电脑计算从而得到蛋白的原子结构[5] [6]。 用NMR研究蛋白质结构的方法,可以在溶液状态进行研究,得到的是蛋白质分子在溶液中的结构,这更接近于蛋白质在生物细胞中的自然状态[7]。此外,通过改变溶液的性质,还可以模拟出生物细胞内的各种生理条件,即蛋白质分子所处的各种环境,以观察这些周围环境的变化对蛋白质分子空间结构的影响。在溶液环境中,蛋白质分子具有与自然环境中类
论述人体组织结构与功能的关系 崔梦梦 (生命科学学院 1241410026) 摘要:结构与功能一直以来不可分割的两个词语,不管是生物还是非生 物,其结构与功能都是息息相关的,要研究功能必然要先分析结构,而 剖析结构时必然会学习其相应的功能。这篇文章从人的肾脏、眼、耳、 肺、小肠这几个部分论述了人体的组织结构与功能的关系。 关键词:结构功能关系肾脏眼耳肺小肠 一、引言 人体的构造很复杂,主要由细胞、组织、器官、系统四级结构组成,每一部分又有其独特的组成与结构,进而形成了其独特的功能,并且各个部分之间相互协调,共同合作,保证人体的正常生活。细胞、组织等的结构是其功能的物质基础,任何一部分的结构发生改变都会影响其功能,进而影响人体的正常运作,而其功能的变化也会影响结构的改变。 二、肾脏的结构与功能的关系 肾实质由大量泌尿小管组成,其间有少量结缔组织、血管和神经等。泌尿小管包括肾小管和集合管两部分。每条肾小管起始端膨大内陷成双层的肾小囊,与肾小球共同构成肾小体,肾小管末端与集合管相连,每个肾小体与一条和它相连的肾小管构成一个肾单位。肾单位是肾的结构与功能的基本单位。每一个肾脏有100万个以上的肾单位。肾单位又可以分为皮质肾单位和髓质肾单位。 肾小管可以分成近端小管,髓袢细段以及远端小管,形成“U”形髓袢,与其相伴行的是“U”形直小血管。肾小管各段以及集合管对水和各种溶质的通透性和重吸收能力不同,因而滤液在流经“U”形髓袢的过程中,由于逆流倍增作用,在肾髓质可造成高渗状态;血液流经“U”形直小血管将水分及部分溶质运走时,由于逆流交换作用,使髓质的高渗状态得以维持;髓袢升支能重吸收溶质而对水不通透,故小管液流到远端小管时一定是低渗的。通过肾的这种结构以及与这种结构相适应的机制可以使机体的尿液维持在一个相对稳定的状态。 肾脏有丰富的血供,正常人两肾的血流量约为每分钟120毫升,相当于心输出量的20%—25%。为全身各脏器灌注量最多的一个。这样大的血流量并非肾代谢所需,而是出于全身血液要求肾及时加工处理以维持内环境稳定的需要。肾脏的血流有90%以上供应肾皮质,仅10%供应肾髓质,肾皮质血流量这么大有利于完成泌尿功能。肾动脉粗而短经多次分支后形成入球小动脉,进入肾小球成为肾
蛋白质结构分析原理及工具 (南京农业大学生命科学学院生命基地111班) 摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。文章侧重于工具的列举,并没有对原理和方法做详细的介绍。文章还列举了蛋白质分析中常用的数据库。 关键词:蛋白质;结构预测;跨膜域;保守结构域 1 蛋白质相似性检测 蛋白质数据库。由一个物种分化而来的不同序列倾向于有相似的结构和功能。物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。表一列出了常用的蛋白质序列数据库和它们的特点。 表一常用蛋白质数据库 网址可能有更新 氨基酸替代模型。进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。氨基酸替代模型可用来估计氨基酸替换的速率。目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。 序列相似性搜索工具。序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。分为局部联配和全局联配。常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。多序列相似性搜索常用于构建系统发育树,这里不阐述。表二列举了常用的成对序列相似性比对搜索工具
转录因子包括什么主要的功能结构域?其主要的结构特点与功能是什么? 作为蛋白质的转录因子从功能上分析其结构可包含有不同区域:①DNA结合域(DNA binding domain),多由60-100个氨基酸残基组成的几个亚区组成;②转录激活域(activating domain),常由30-100氨基酸残基组成,这结构域有富含酸性氨基酸、富含谷氨酰胺、富含脯氨酸等不同种类,一酸性结构域最多见; ③连接区,即连接上两个结构域的部分。不与DNA直接结合的转录因子没有DNA 结合域,但能通过转录激活域直接或间接作用与转录复合体而影响转录效率。 与DNA结合的转录因子大多以二聚体形式起作用,与DNA结合的功能域常见有以几种: ①螺旋-转角-螺旋(helix-turn-helix,HTH)及螺旋-环-螺旋(helix-loop-helix,HLH) 这类结构至少有两个α螺旋其间由短肽段形成的转角或环连接,两个这样的motif结构以二聚体形式相连,距离正好相当于DNA一个螺距(3.4nm),两个α螺旋刚好分别嵌入DNA的深沟。 ②锌指(zinc finger)其结构如图所示,每个重复的“指”状结构约含23个氨基酸残基,锌以4个配价键与4个半胱氨酸、或2个半胱氨酸和2个组氨酸相结合。整个蛋白质分子可有2-9个这样的锌指重复单位。每一个单位可以其指部伸入DNA双螺旋的深沟,接触5个核苷酸。例如与GC盒结合的转录因子SP1 中就有连续的3个锌指重复结构。 ③碱性-亮氨酸拉链(basic leucine zipper,bZIP)这结构的特点是蛋白质分子的肽链上每隔6个氨基酸就有一个亮氨酸残基,结果就导致这些亮氨酸残基都在α螺旋的同一个方向出现。两个相同的结构的两排亮氨酸残基就能以疏水键结合成二聚体,这二聚体的另一端的肽段富含碱性氨基酸残基,借其正电荷与DNA 双螺旋链上带负电荷的磷酸基团结合。若不形成二聚体则对DNA的亲和结合力明显降低。在肝脏、小肠上皮、脂肪细胞和某些脑细胞中有称为C/EBP家族的一大类蛋白质能够与CAAT盒和病毒增强子结合,其特征就是能形成bZIP二聚体结构。
分析蛋白结构域(Domains)的三种方法 生物信息编程2009-09-24 23:55:50 阅读1235 评论0 字号:大中小订阅 三种分析蛋白结构域(Domains)的方法 1,SMART入门,蛋白结构和功能分析 SMART介绍 SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART. SMART(http://smart.embl-heidelberg.de/),可以说是蛋白结构预测和功能分析的工具集合。简单点说,就是集合了一些工具,可以预测蛋白的一些二级结构。如跨膜区(Transmembrane segments),复合螺旋区(coiled coil regions),信号肽(Signal peptides),蛋白结构域(PFAM domains)等。 SMART前该知道的 1,SMART有两种不同的模式:normal 或genomic 主要是用的数据库不一样。Normal SMART, 用的数据库Swiss-Prot, SP-TrEMBL 和stable Ensembl proteomes。Genomic SMART, 用全基因组序列。详细列表:http://smart.embl-heidelberg.de/smart/list_genomes.pl 2,一些名词解释 http://smart.embl-heidelberg.de/help/smart_glossary.shtml
第六章 蛋白质的功能域、结构及其药物设计 随着人类基因组全序列测定的完成,预示着基因组研究从结构基因组(Structural Genomics)进入了功能基因组(Functional Genomics)研究时代。研究基因组功能当然首先要研究基因表达的模式。当前研究这一问题可以基于核酸技术,也可以基于蛋白质技术,即直接研究基因的表达产物。测定一个有机体的基因组所表达的全部蛋白质的设想是由Williams于1994年正式提出的,而“蛋白质组”(proteome)一词是Wilkins于1995年首次提出。蛋白质组是指由一个细胞或组织的基因组所表达的全部相应的蛋白质。蛋白质组与基因组相对应,均是一个整体概念,但是两者又有根本的不同:一个有机体只有一个确定的基因组,组成该有机体的所有不同细胞都共享有一个基因组;但是,基因组内各个基因表达的条件、时间和部位等不同,因而它们的表达产物(蛋白质)也随条件、时间和部位的不同而有所不同。因此,蛋白质组又是一个动态的概念。由于以上原因,再加上由于基因剪接,蛋白质翻译后修饰和蛋白质剪接,基因遗传信息的表达规律更趋复杂,不再是经典的一个基因一个蛋白的对应关系,而是一个基因可以表达的蛋白质数目大于一。由此可见,蛋白质组研究是一项复杂而艰巨的任务。 蛋白质结构与功能的研究已有相当长的历史,由于其复杂性,对其结构与功能的预测不论是方法论还是基础理论方面均较复杂。统计学方法曾被成功地应用于蛋白质二级结构预测中,如Chou和Fasman提出的经验参数法便是最突出的例子。 该方法统计分析了各种氨基酸的二级结构分布特征,得出相应参数(P а,P β 和P t )并 用于预测。本章将简要介绍蛋白质结构与功能预测的生物信息学途径。 第一节 蛋白质功能预测 一、根据序列预测功能的一般过程 如果序列重叠群(contig)包含有蛋白质编码区,则接下来的分析任务是确定表达产物——蛋白质的功能。蛋白质的许多特性可直接从序列上分析获得,如疏水性,它可以用于预测序列是否跨膜螺旋(transmenbrane helix)或是前导序列(leader sequence)。但是,总的来说,我们根据序列预测蛋白质功能的唯一方法是通过数据库搜寻,比较该蛋白是否与已知功能的蛋白质相似。有2条主要途径可以进行上述的比较分析: ①比较未知蛋白序列与已知蛋白质序列的相似性; ②查找未知蛋白中是否包含与特定蛋白质家族或功能域有关的亚序列或保守区段。 图6.1给出了根据序列预测蛋白质功能的大致过程。由于涉及数条技术路线,所得出的分析结果并不会总是相一致。一般来说,数据库相似性搜索获得的结果最为可靠,而来自PROSITE的结果相对不可靠。
1蛋白质家族和结构域数据库 1.1蛋白质模体及结构域数据库 模体和结构域 PROSITE数据库 PRINTS数据库 BLOCKS数据库 ProDom数据库 Pfam数据库 SMART数据库 InterPro数据库 Conserved Domain数据库 CDART 模体(motifs)和结构域(domains): Biologists can gain insight of the protein function based on identification of short consensus sequences related to known functions. These consensus sequence patterns are termed motifs and domains. A motif is a short conserved sequence pattern associated with distinct functions of a protein or DNA. It is often associated with a distinct structural site performing a particular function. A typical motif, such as a Zn-finger motif, is ten to twenty amino acids long. A domain is also a conserved sequence pattern, defined as an independent functional and structural unit. Domains are normally longer than motifs. A domain consists of more than 40 residues and up to 700 residues, with an average length of 100 residues. A domain may or may not include motifs within its boundaries. Examples,transmembrane domains, ligand-binding domains. Identification of motifs and domains heavily relies on multiple sequence alignment as well as profile and hidden Markov model (HMM) construction PROSITE(蛋白质家族及结构域数据库): The first established sequence pattern database https://www.doczj.com/doc/378548622.html,/prosite/ 是蛋白质家族和结构域数据库,包含具有生物学意义的位点、模式、可帮助识别蛋白质家族的统计特征。 PROSITE中涉及的序列模式包括酶的催化位点、配体结合位点、与金属离子结合的残基、二硫键的半胱氨酸、与小分子或其它蛋白质结合的区域等。 PROSITE还包括根据多序列比对而构建的序列统计特征,能更敏感地发现一个(未知)序列是否具有相应的特征。 The functional information of these patterns is primarily based on published literature. PRINTS(蛋白质模体指纹数据库):
蛋白质序列、性质、功能和结构分析 基于网络的蛋白质序列检索与核酸类似,从NCBI或利用SRS系统从EMBL检索。 1、疏水性分析ExPASy的ProtScale程序(https://www.doczj.com/doc/378548622.html,/cgi-bin/protscale.pl)可用来计算蛋白质的疏水性图谱。输入的数据可为蛋白质序列或SWISS-PROT数据库的序列接受号。也可用BioEdit、DNAMAN等软件进行分析。 2、跨膜区分析蛋白质跨膜区域分析的网络资源有: TMPRED:https://www.doczj.com/doc/378548622.html,/software/TMPRED_form.html PHDhtm: http:www.embl-heidelberg.de/Services/ ... predictprotein.html MEMSAT: ftp://https://www.doczj.com/doc/378548622.html, 3、前导肽和蛋白质定位一般认为,蛋白质定位的信息存在于该蛋白自身结构中,并且通过与膜上特殊受体的相互作用得以表达。这就是信号肽假说的基础。这一假说认为,穿膜蛋白质是由 mRNA编码的。在起始密码子后,有一段疏水性氨基酸序列的RNA片段,这个氨基酸序列就称为信号序列(signal sequence)。蛋白质序列的信号肽分析可联网到http://genome.cbs.dtu.dk/services/SignalP/或其二版网址http: //genome.cbs.dtu.dk/services/SignalP-2.0/。该服务器也提供利用e-mail 进行批量蛋白质序列信号肽分析的方案(http://genome.cbs.dtu.dk/services/SignalP/mailserver.html),e-mail 地址为 signalp@ genome.cbs.dtu.dk。蛋白质序列中含有的信号肽序列将有助于它们向细胞内特定区域的移动,如前导肽和面向特定细胞器的靶向肽。在线粒体蛋白质的跨膜运输过程中,通过线粒体膜的蛋白质在转运之前大多数以前体形式存在,它由成熟蛋白质和N端延伸出的一段前导肽或引肽(leader peptide)共同组成。迄今有40多种线粒体蛋白质前导肽的一级结构被阐明,它们约含有20~80个氨基酸残基,当前体蛋白跨膜时,前导肽被一种或两种多肽酶所水解转变成成熟蛋白质,同时失去继续跨膜能力。前导肽一般具有如下性质:①带正电荷的碱性氨基酸(特别是精氨酸)含量较丰富,它们分散于不带电荷的氨基酸序列中间;②缺失带负电荷的酸性氨基酸;③羟基氨基酸(特别是丝氨酸)含量较高;④有形成两亲(即有亲水又有疏水部分)α-螺旋结构的能力。和信号肽与跨膜区结构一样,蛋白质的亚细胞定位也和其功能密切相关,蛋白质亚细胞定位分析可通过如下网址进行:http://predict.
4.2 针对蛋白质的预测方法 传统的生物学认为,蛋白质的序列决定了它的三维结构,也就决定了它的功能。由于用X光晶体衍射和NMR核磁共振技术测定蛋白质的三维结构,以及用生化方法研究蛋白质的功能效率不高,无法适应蛋白质序列数量飞速增长的需要,因此近几十年来许多科学家致力于研究用理论计算的方法预测蛋白质的三维结构和功能,经过多年努力取得了一定的成果。 1. 从氨基酸组成辨识蛋白质 根据组成蛋白质的20种氨基酸的物理和化学性质可以分析电泳等实验中的未知蛋白质,也可以分析已知蛋白质的物化性质。ExPASy工具包中提供了一系列相应程序: AACompIdent:根据氨基酸组成辨识蛋白质。这个程序需要的信息包括:氨基酸组成、蛋白质的名称(在结果中有用)、pI和Mw(如果已知)以及它们的估算误差、所属物种或物种种类或“全部(ALL)”、标准蛋白的氨基酸组成、标准蛋白的SWISS-PROT编号、用户的Email地址等,其中一些信息可以没有。这个程序在SWISS-PROT和(或)TrEMBL数据库中搜索组成相似蛋白。 AACompSim:与前者类似,但比较在SWISS-PROT条目之间进行。这个程序可以用于发现蛋白质之间较弱的相似关系。 除了ExPASy中的工具外,PROPSEARCH也提供基于氨基酸组成的蛋白质辨识功能。程序作者用144种不同的物化性质来分析蛋白质,包括分子量、巨大残基的含量、平均疏水性、平均电荷等,把查询序列的这些属性构成的“查询向量”与SWISS-PROT和PIR中预先计算好的各个已知蛋白质的属性向量进行比较。这个工具能有效的发现同一蛋白质家族的成员。可以通过Web使用这个工具,用户只需输入查询序列本身。 ExPASy的网址是:http://www.expasy.ch/tools/。 PROSEARCH的网址是:http://www.embl-heidelberg.de/prs.html。 2. 预测蛋白质的物理性质 从蛋白质序列出发,可以预测出蛋白质的许多物理性质,包括等电点、分子量、酶切特性、疏水性、电荷分布等。相关工具有: Compute pI/MW:是ExPASy工具包中的程序,计算蛋白质的等电点和分子量。对于碱性蛋白质,计算出的等电点可能不准确。 PeptideMass:是ExPASy工具包中的程序,分析蛋白质在各种蛋白酶和化学试剂处理后的内切产物。蛋白酶和化学试剂包括胰蛋白酶、糜蛋白酶、LysC、溴化氰、ArgC、AspN 和GluC等。
蛋白结构分析和比较 姓名________ 学号______________ 日期________年___月___日 阅读分子月报科普短文,参阅相关文献,从蛋白质结构数据库下载以下蛋白质三维结构原子坐标文件,利用Swiss-PdbViewer显示观察,说明其结构特点。 猪胰岛素(4INS): 由几个亚基组成,每个亚基有几条多肽链,每条多肽链由哪些二级结构单元组成; 每条多肽链有几对链内二硫键,多肽链之间由几对二硫键连接; 每个亚基如何与锌原子结合。 抹香鲸肌红蛋白(1MBO): 由几股alpha螺旋组成; 与血色素卟啉环中央铁原子以配位健结合的是哪个组氨酸,该组氨酸位于第几股alpha 螺旋; 与血色素携带的氧分子通过氢键连接的是哪个组氨酸,该组氨酸位于第几股alpha螺旋。 小鼠免疫球蛋白(1IGT): 由几个亚基组成,每个亚基各有几个结构域; 两条重链之间由几对二硫键连接,重链和轻链之间由几对二硫键连接; 每个结构域内部的二硫键和色氨酸如何形成疏水内核; 多糖链对稳定分子结构的作用。 水母(Jellyfish)绿色荧光蛋白(1GFL): 选择PDB原始文件中二聚体A链,保存为单个亚基1GFLa.pdb; 打开1GFLa.pdb,并用不同颜色显示二级结构beta折叠; 找出分子内部发光基团Ser65-Tyr66-Gly67并说明其发光机理。 核小体(1AOI): 用不同颜色显示组蛋白8个亚基; 观察DNA分子碱基配对特点; 显示组蛋白表面与DNA相互作用的碱性氨基酸。 斑头雁和灰雁血红蛋白比较实例 从UniProt数据库中提取斑头雁和灰雁血红蛋白alpha亚基序列,进行序列比对,找出差异位点。 用SwissPDB-Viwer软件中选择并保存灰雁氧合血红蛋白1FAW中四个亚基中的A链B 链两个亚基。 用结构叠合方法分析比较灰雁氧合血红蛋白A链B链两个亚基与斑头雁血红蛋白1A4F 两个亚基的结构,计算基于alpha碳叠合后的均方根误差(RMSD)。 找出斑头雁血红蛋白A链第119位丙氨酸侧链beta碳原子CB和B链55位亮氨酸侧链末端两个碳原子CD1和CD2,分别测量A119CB和B55CD1、B55CD2之间的距离。 找出灰雁血红蛋白A链第119位脯氨酸侧链gamma碳原子CG和B链55位亮氨酸侧链末端两个碳原子CD1和CD2,分别测量A119CG和B55CD1、B55CD2之间的距离。 根据上述分析结果,参阅相关文献,说明斑头雁和灰雁血红蛋白A119侧链大小和柔性不同,如何影响其构象变化,从而进一步引起氧气结合能力的变化。 利用模拟突变的方法,将灰雁血红蛋白A链第119位脯氨酸突变成丙氨酸,测量突变后的A119CB和B55CD1、B55CD2之间的距离。 课题相关蛋白质结构分析
蛋白质结构与功能的关系 专业:植物学 摘要:蛋白质特定的功能都是由其特定的构象所决定的,各种蛋白质特定的构象又与其一级结构密切相关。天然蛋白质的构象一旦发生变化,必然会影响到它的生物活性。由于蛋白质的构象的变化引起蛋白质功能变化,可能导致蛋白质构象紊乱症,当然也能引起生物体对环境的适应性增强。而分子模拟技术为蛋白质的研究提供了一种崭新的手段。在理论上解决了结构预测和功能分析以及蛋白质工程实施方面所面临的难题。它在蛋白质的结构预测和模建工作中占有举足轻重的地位,实现了生物技术与计算机技术的完美结合。 关键词:蛋白质的结构、功能;折叠/功能关系;蛋白质构象紊乱症;分子模拟技术;同源建模 RNase是由124个氨基酸残基组成的单肽链,分子中 8 个Cys的-SH构成4对二硫键,形成具有一定空间构象的蛋白质分子。在蛋白质变性剂和一些还原剂存在下,酶分子中的二硫键全部被还原,酶的空间结构破坏,肽链完全伸展,酶的催化活性完全丧失。当用透析的方法除去变性剂和巯基乙醇后,发现酶大部分活性恢复,所有的二硫键准确无误地恢复原来状态。若用其他的方法改变分子中二硫键的配对方式,酶完全丧失活性。这个实验表明,蛋白质的一级结构决定它的空间结构,而特定的空间结构是蛋白质具有生物活性的保证。前体与活性蛋白质一级结构的关系,由108个氨基酸残基构成的前胰岛素原,在合成的时候完全没有活性,当切去N-端的24个氨基酸信号肽,形成84个氨基酸的胰岛素原,胰岛素原也没活性,在包装分泌时,A、B链之间的33个氨基酸残基被切除,才形成具有活性的胰岛素。 功能不同的蛋白质总是有着不同的序列;种属来源不同而功能相同的蛋白质的一级结构,可能有某些差异,但与功能相关的结构也总是相同。若一级结构变化,蛋白质的功能可能发生很大的变化。蛋白质特定的功能都是由其特定的构象所决定的,各种蛋白质特定的构象又与其一级结构密切相关。天然蛋白质的构象一旦发生变化,必然会影响到它的生物活性。由于蛋白质的构象的变化引起蛋白质功能变化,可能导致蛋白质构象紊乱症,当然也能引起生物体对环境的适应性增强。 虽然蛋白质结构与生物功能的关系比序列与功能的关系更加紧密,但结构与功能的这种关联亦若隐若现,并不能排除折叠差别悬殊的蛋白质执行相似的功能,折叠相似的蛋白质执行差别悬殊功能的现象的存在。无奈,该领域仍不得不将100多年前Fisher提出的“锁一钥
实验名称:蛋白质结构与功能的生物信息学研究实验目的:1.掌握运用BLAST工具对指定蛋白质的氨基酸序列同源性搜索 的方法。 2.掌握用不同的工具分析蛋白质的氨基酸序列的基本性质 3掌握蛋白质的氨基酸序列进行三维结构的分析 4.熟悉对蛋白质的氨基酸序列所代表蛋白的修饰情况、所参与的 代谢途径、相互作用的蛋白,以及与疾病的相关性的分析。 实验方法和流程: 一、同源性搜索 同源性从分子水平讲则是指两个核酸分子的核苷酸序列或两个蛋白质分子的氨基酸序列间的相似程度。BLAST工具能对生物不同蛋白质的氨基酸序列或不同的基因的DNA序列极性比对,并从相应数据库中找到相同或相似序列。对 指定的蛋白质的氨基酸序列进行同源性搜索步骤如下: ↓ 登录网址https://www.doczj.com/doc/378548622.html,/blast/ ↓ 输入序列后,运行blast工具 ↓ 序列比对的图形结果显示
序列比对的图形结果:用相似性区段(Hit)覆盖输入序列的范围判断两个序列 的相似性。如果图形中包含低得分的颜色(主要是红色) 区段,表明两序列的并非完全匹配。 ↓ 匹配序列列表及得分
各序列得分 可选择不同的比对工具 备注: Clustal是一款用来对()的软件。可以用来发现特征序列,进行蛋白分类,证明 序列间的同源性,帮助预测新序列二级结构与三级结构,确定PCR引物,以及 在分子进化分析方面均有很大帮助。Clustal包括Clustalx和Clustalw(前者是图 形化界面版本后者是命令界面),是生物信息学常用的多序列比对工具。 该序列的比对结果有100条,按得分降序排列,其中最大得分2373,最小得分 分为1195. ↓ 详细的比对序列的排列情况 第一个匹配 序列 第一个序列的匹配率为100% Score表示打分矩阵计算出来的值,由搜索算法决定的,值越大说明匹配程度
第一章蛋白质的结构与功能 [测试题] 一、名词解释:1.氨基酸 2.肽 3.肽键 4.肽键平面 5.蛋白质一级结构 6.α-螺旋 7.模序 8.次级键 9.结构域 10.亚基 11.协同效应 12.蛋白质等电点 13.蛋白质的变性 14.蛋白质的沉淀 15.电泳 16.透析 17.层析 18.沉降系数 19.双缩脲反应 20.谷胱甘肽 二、填空题 21.在各种蛋白质分子中,含量比较相近的元素是____,测得某蛋白质样品含氮量为15.2克,该样品白质含量应为____克。 22.组成蛋白质的基本单位是____,它们的结构均为____,它们之间靠____键彼此连接而形成的物质称为____。 23.由于氨基酸既含有碱性的氨基和酸性的羧基,可以在酸性溶液中带____电荷,在碱性溶液中带____电荷,因此,氨基酸是____电解质。当所带的正、负电荷相等时,氨基酸成为____离子,此时溶液的pH值称为该氨基酸的____。 24.决定蛋白质的空间构象和生物学功能的是蛋白质的____级结构,该结构是指多肽链中____的排列顺序。25.蛋白质的二级结构是蛋白质分子中某一段肽链的____构象,多肽链的折叠盘绕是以____为基础的,常见的二级结构形式包括____,____,____和____。 26.维持蛋白质二级结构的化学键是____,它们是在肽键平面上的____和____之间形成。 27.稳定蛋白质三级结构的次级键包括____,____,____和____等。 28.构成蛋白质的氨基酸有____种,除____外都有旋光性。其中碱性氨基酸有____,____,____。酸性氨基酸有____,____。 29.电泳法分离蛋白质主要根据在某一pH值条件下,蛋白质所带的净电荷____而达到分离的目的,还和蛋白质的____及____有一定关系。 30.蛋白质在pI时以____离子的形式存在,在pH>pI的溶液中,大部分以____离子形式存在,在pH
相关主题
文本预览