血液DNA快速提取方法
- 格式:doc
- 大小:19.50 KB
- 文档页数:2

磁珠法全血基因组DNA 提取详细步骤及方法注意事项样品应避免反复冻融,否则会导致提取的DNA片段较小且提取量也下降;◆需要自备材料:无菌水、异丙醇、磁铁或磁架、1.5 ml离心管(最好低吸附的离心管);需要去除RNA自备RNase A,在蛋白酶K消化这一步加入4 μl RNase A (100 mg/ml);PBS。
◆已加入抗凝剂的血液样品可在2-8℃储存不超过3天(推荐使用EDTA作为抗凝剂),长期贮存请置于-70℃;◆整个过程请勿离心,以免磁珠不可逆聚集而影响提取效果。
实验准备◆冻存血液应在室温或37℃缓慢解冻,不可高温加热,以免血凝成块;将冻存的血液置于37℃摇床150-200 rpm融化,效果最佳;也可提前一天放在4℃解冻;◆磁珠用前一定要充分混匀;◆56℃加热源◆使用前需在Buffer BGW I中加入相应体积的无水乙醇。
操作步骤1. 裂解:(1)当全血样品量低于200 μl时,在全血中加入200 μl Buffer BGL、20 μl Proteinase K,56℃ 10min;(2)当全血样品量高于200 μl时,12000 rpm离心1分钟,弃上清,加入200 μl PBS缓冲液,再加入200 μl Buffer BGL、20 μl Proteinase K,56℃ 10min;2. 结合:在上述裂解液中加入400 μl 异丙醇和磁珠悬浮液(MB) 30 μl(使用前充分重悬),吹打分散磁珠;将离心管放置于磁力架上,待磁珠完全吸附后吸弃液体。
3.漂洗: (1) 将离心管从磁力架上取下,向离心管中加入500 μl Buffer BGW 0,涡旋振荡5s,充分重悬磁珠,将离心管放置于磁力架上,待磁珠完全吸附后吸弃液体,重复该步骤一次。
(2) 将离心管从磁力架上取下,向离心管中加入500 μl Buffer BGW I,涡旋振荡5s,充分重悬磁珠,将离心管放置于磁力架上,待磁珠完全吸附后吸弃液体。

提取dna的实验步骤原理DNA提取是一项重要的实验技术,用于从细胞中分离纯净的DNA。
它在生物学、医学、犯罪学等领域具有广泛的应用。
DNA提取的过程可以分为以下几个步骤:样品收集、细胞破碎、蛋白质消化、DNA分离、洗涤、溶解和纯化。
首先,样品收集是DNA提取的第一步。
样品可以来自不同的来源,如血液、唾液、组织样本等。
对于血液样品,静脉采血是最常见的方式。
收集的血液样品需要采用抗凝剂进行预处理,以防止凝血。
细胞破碎是DNA提取的关键步骤之一。
细胞破碎的目的是将细胞膜以及核膜破坏,释放细胞内的DNA。
常用的细胞破碎方法有物理破碎、化学破碎和酶解法。
物理破碎一般采用高速离心仪或超声波处理,通过机械力量破坏细胞结构。
化学破碎使用适当的溶液,如细胞裂解液(例如EDTA、SDS、蛋白酶K等)来分解膜脂质和蛋白质。
酶解法则通过酶的作用来降解膜和核酸。
蛋白质消化是为了去除提取过程中的蛋白质杂质。
蛋白质会干扰DNA的纯化和测定。
在蛋白质消化过程中,常用的方法是加入蛋白酶K或肾脏酸性蛋白酶等蛋白水解酶,通过其酶解作用将蛋白质降解。
DNA分离是将DNA与其他细胞组分分离的过程。
DNA在细胞破碎后呈现为线性或环状的形态。
为了分离DNA,一般采用盐溶液和有机溶剂共同作用的方案,如添加高浓度的盐类(如NaCl)和异丙醇。
这些溶剂会沉淀细胞残渣和蛋白质,而DNA则溶于水相中。
洗涤过程是为了去除残留的蛋白质和盐类。
洗涤液一般为酒精溶液,如酒精和醋酸盐。
洗涤时,将沉淀的DNA加入洗涤液中,通过离心使DNA沉淀,然后去除上清液。
此过程重复一至三次可以有效去除杂质。
溶解是将沉淀的DNA重新溶解于缓冲液中,以便于后续的操作。
溶解液可以是Tris-EDTA缓冲液、无菌纯水等。
在加入缓冲液后,将溶液置于水浴中进行搅拌和温度控制,使DNA彻底溶解。
纯化是最后一步,目的是去除DNA提取过程中的杂质,获得纯净的DNA。
常见的纯化方法有酚-氯仿抽提法和硅胶纯化法。

1.-20度冻存1年是没有问题的,当然越低越好。
但是冻存后,用蛋白酶K法,红细胞不易裂解,必须采用特殊方法裂解。
2.建议裂解红细胞后再冻存于-80度。
因为冻融后红细胞会裂解,而且红细胞会增加蛋白污染,影响DNA抽提3.我们实验室加ACD抗凝血液,保存3年了,抽提DNA没问题(我们是先离心后分层保存,以便后续试验)!血DNA提取技术分离外周血白细胞提取方法:试验步骤:1、取人肘静脉血5ml,EDTA抗凝,2500rpm离心10min。
2、小心吸取上层血浆,分装到3个0.5ml离心管中。
3、在血细胞中加入3倍体积的溶血液,摇匀,冰浴15min。
4、2500rpm离心10min,弃上清。
5、加入10ml溶血液,摇匀,冰浴15min。
6、3000rpm离心10min,弃上清。
7、倒置离心管,去掉残液。
8、得白细胞,-80?C冻存。
试验要求:血至分离白细胞之间隔时间在室温下放置不超过2h,4℃放置不超过5h,以防白细胞自溶。
氯仿法抽提外周血白细胞基因组DNA:试验试剂:Ligsisbuffer:133mMNH4ClNHCl7.12g0.9mMNH4HCO3NH4HCO30.071g0.1mMEDTA0.5mMEDTA0.2ml;最后加灭菌去离子水至1000ml,高压灭菌。
ACD抗凝剂:柠檬酸1.68g柠檬酸钠4.62g葡萄糖5.15g;最后加灭菌去离子水至350ml,高压灭菌。
提取缓冲液(Extractionbuffer):10mMTrisCl(PH=8.0)1MTris.Cl(PH=8.0)1ml0.1mMEDTA(PH=8.0)0.5mMEDTA(PH=8.0)20ml0 .5%SDS10%SDS0.5ml;最后加灭菌去离子水至100ml,高压灭菌试验步骤:1、在500μl抗凝血中加入ligsisbuffer1000μl,充分颠混至清亮。
以4000rpm,离心5min。
弃上清液。
2、沉淀中加入ligsisbuffer1500μl,充分匀浆。

磁珠法提取dna原理磁珠法提取DNA原理DNA提取是分子生物学研究中的重要步骤,磁珠法是一种常用的DNA提取方法。
磁珠法利用了磁性珠子的特性,使得DNA在磁场作用下能够与磁珠结合,从而实现DNA的快速、高效提取。
磁珠法提取DNA的原理基于亲和层析技术,其中亲和剂是磁性珠子表面的修饰物。
这些修饰物能够与DNA的特定区域结合,例如特定序列、结构或染色体上的特定区域。
磁珠法的基本步骤包括样品裂解、DNA与磁珠结合、磁珠分离、洗涤和DNA的洗脱。
样品需要经过裂解步骤,以破坏细胞膜和核膜,使DNA释放到溶液中。
裂解液中通常包含蛋白酶K和蛋白酶K缓冲液,以消化细胞蛋白质。
然后,磁性珠子被加入裂解液中,这些珠子的表面修饰物能够与DNA结合。
修饰物可以是亲和剂,如亲合素或抗体,也可以是特定的DNA引物。
在结合步骤中,磁性珠子与DNA结合形成复合物。
通过磁场的作用,磁珠复合物被吸附到管壁或磁珠法专用的磁力架上,从而实现DNA的分离。
与传统的离心分离方法相比,磁珠法具有更高的纯度和回收率。
此外,磁珠复合物的形成和分离速度较快,节省了实验时间。
分离步骤通常涉及将磁力架移除至洗涤液中,以去除非特异性结合物质。
洗涤步骤可以使用乙酸盐缓冲液、乙醇或其他洗涤缓冲液来去除冗余的污染物。
洗涤后,磁力架被重新放置到洗脱液中,DNA 从磁珠上解离,溶解在洗脱液中。
洗脱液通常是低盐缓冲液或去离子水,以确保DNA的高纯度。
磁珠法提取DNA的优点不仅在于其高纯度和回收率,还在于其灵活性和可扩展性。
磁珠的表面修饰物可以根据实验需求进行选择,以实现对特定DNA序列或特定组分的选择性结合。
此外,磁珠法可应用于多种样品类型,包括血液、组织、细胞和体液等。
对于大规模的DNA提取,磁珠法可以进行高通量自动化处理,提高操作效率和样品数量。
总结起来,磁珠法提取DNA的原理是利用磁性珠子与DNA的特异性结合,通过磁场的作用将DNA分离和纯化。
这种方法具有高纯度、高回收率、灵活性和可扩展性的优点,广泛应用于分子生物学研究、临床诊断和法医学等领域。


法医实验室几种常用DNA提取方法及比较法医实验室几种常用DNA提取方法及比较(一)Chelex100法原理:Chelex100是一种螯合树脂,由苯乙烯、二乙烯苯共聚体组成,含有成对的亚氨基二乙酸盐离子,起着螯合基团作用,对多价金属离子有极强亲和力。
在低离子强度、碱性及100℃煮沸条件下,可以使细胞膜裂解,并使与DNA结合的蛋白质变性,DNA游离。
检材基质中一般含有大量金属离子,在低离子强度及加热条件下,金属离子可以辅助脱氧核糖核酸酶降解DNA,也可以抑制PCR反应,所以在提取DNA时,加入Chelex100,螯合了金属离子,防止了DNA降解,提高了PCR扩增成功率。
方法:剪取适量血斑、精斑、汗斑、鼻涕斑、指甲、毛根、软组织等等生物检材,置于0.5ml离心管内,加入适量纯水,室温浸泡,13,000rpm离心5min,上清丢弃,管底留约20μl左右液体及检材基质,加入100-200μl 5%Chelex100(有时需加适量PK)56℃15mi n至数10小时不等,100℃8min,13,000rpm离心5mim,上清备用。
评述:1、Chelex100法已经成为DNA提取的基本方法,Chelex100法是万能的。
目前,我们一切纯化方法都在Chelex100法的基础上进行,Chelex100法又不是万能的。
2、Chelex100法提取前的检材清洗非常重要,因为现场检材往往均较脏,脏东西可以抑制PCR反应,所以往往不止清洗一次,清洗检材时可能损失部分DNA。
所以应平衡清洗抑制剂与损失DNA之间的关系。
特别强调清洗时应“把根留住”,很多情况下已知样本DNA检验失败的原因是把根没有留住。
DNA检验时还有另外一句名言:“一个萝卜一个坑”,防止混乱检材。
肝素抑制PCR反应,血红素抑制PCR反应,所以长时的浸泡、多次清洗往往可能洗除肝素及血红素。
在已知样本多次无检验结果疑为肝素抗凝的情况下,多洗是检验成功所必须的。
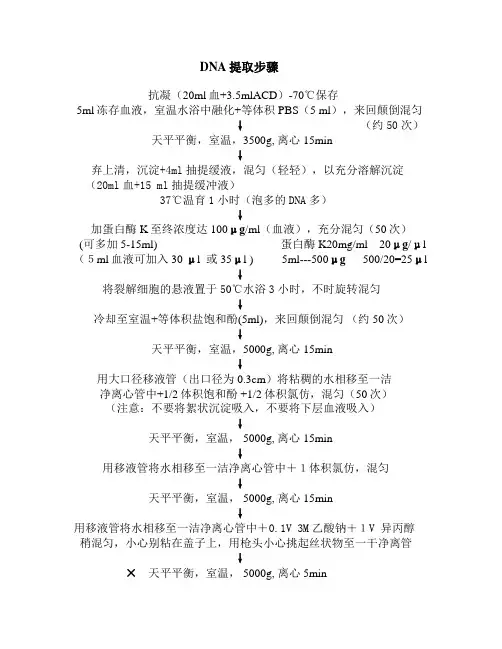
DNA提取步骤抗凝(20ml血+3.5mlACD)-70℃保存5ml冻存血液,室温水浴中融化+等体积PBS(5 ml),来回颠倒混匀↓(约50次)天平平衡,室温,3500g, 离心15min↓弃上清,沉淀+4ml抽提缓液,混匀(轻轻),以充分溶解沉淀(20ml血+15 ml抽提缓冲液)37℃温育1小时(泡多的DNA多)↓加蛋白酶K至终浓度达100μg/ml(血液),充分混匀(50次)(可多加5-15ml)蛋白酶K20mg/ml20μg/μl (5ml血液可加入30 μl 或35μl ) 5ml---500μg 500/20=25μl↓将裂解细胞的悬液置于50℃水浴3小时,不时旋转混匀↓冷却至室温+等体积盐饱和酚(5ml),来回颠倒混匀(约50次)↓天平平衡,室温,5000g, 离心15min↓用大口径移液管(出口径为0.3cm)将粘稠的水相移至一洁净离心管中+1/2体积饱和酚 +1/2体积氯仿,混匀(50次)(注意:不要将絮状沉淀吸入,不要将下层血液吸入)↓天平平衡,室温, 5000g, 离心15min↓用移液管将水相移至一洁净离心管中+1体积氯仿,混匀↓天平平衡,室温, 5000g, 离心15min↓用移液管将水相移至一洁净离心管中+0.1V 3M乙酸钠+1V 异丙醇稍混匀,小心别粘在盖子上,用枪头小心挑起丝状物至一干净离管↓×天平平衡,室温, 5000g, 离心 5min↓沉淀用20%乙醇洗两次,收集DNA(多洗几次,把DNA放在壁上,用枪头吸干乙醇洗4次,无须离心)↓晾干乙醇+20-50μl TE液 100μl TE液(溶解DNA)↓摇床12-24小时,直至DNA完全溶解,如隔几天使用,无须摇床,DNA也能完全溶解↓测定DNA样品在260nm和280nm处的光吸收值,计算DNA纯度和浓度↓DNA于4℃下保存。


鱼类血液基因组DNA的提取1.取0.3ml血液于1.5ml离心管中,加入1/6体积(0.05ml)ACD抗凝剂,再加入1ml 0.35 %NaCl 淡水鱼生理盐溶液, 混匀,12 000 r/ min离心2 min (若上清仍带红色可加入0.35 % NaCl 再洗一次) 去上清;2.沉淀加入100μl 0.35 %NaCl 摇匀, 加入400μl 无离子水作低渗处理15 min , 13 000 r/min离心3 min , 去上清, 获乳白色沉淀( 细胞核) ;3.向沉淀中加入100 μl0135 % NaCl , 混匀, 再加入400μl 裂解液缓缓混匀后, 加入蛋白酶K (20mg/ml) 10μl , 温和混匀,置入50 ℃水浴5 h。
4.将消化的样品加入等体积的Tris饱和酚,充分摇匀,12000rpm离心10min;5.将上层水相转入另一无菌1.5ml离心管中,加入等体积的酚∶氯仿∶异戊醇(25∶24∶1),旋涡振荡至充分混匀,12000rpm离心10min;6.将上层水相转入另一无菌1.5ml离心管中;并加入等体积的氯仿∶异戊醇(24∶1),充分振荡混匀,12000rpm离心10min;7.将上清液转移到另一无菌1.5ml离心管中;加入2倍体积的冰乙醇,1/10体积醋酸钠(NaAc)水平摇动,直到可见絮状DNA析出;8.将样品置-20℃冰箱冷冻30min或冰浴15~20min,取出后再次12000rpm离心10min,使DNA沉淀;9.倒去上清液,把离心管倒扣在吸水纸上,吸干液体。
用700μl 70%乙醇洗涤并晃动,以洗出DNA的杂质;10.倒出乙醇,将DNA用真空干燥或自然晾干,加入适量的TE缓冲液或无菌水回溶,-20℃保存备用。

血液dna 提取流程英文回答:Blood DNA Extraction Protocol.Materials:Whole blood sample in EDTA tube.DNA extraction kit.Microcentrifuge.Pipettes and tips.Vortex mixer.Water bath or heating block.Ethanol (96-100%)。
RNase A.Procedure:1. Lysis and Proteinase K Digestion.Add 200 µL of whole blood to a microcentrifuge tube.Add 20 µL of proteinase K solution and mix thoroughly.Incubate at 56°C for 10 minutes.2. DNA Binding.Add 200 µL of lysis buffer and vortex vigorously.Add 200 µL of ethanol (96-100%) and mix thoroughly.Pipette the mixture onto a spin column and centrifugeat 14,000 x g for 1 minute.3. Washing.Add 500 µL of wash buffer 1 and centrifuge at 14,000x g for 1 minute.Add 500 µL of wash buffer 2 and centrifuge at 14,000x g for 1 minute.4. Elution.Place the spin column in a clean microcentrifuge tube.Add 50-100 µL of elution buffer and incubate at room temperature for 5 minutes.Centrifuge at 14,000 x g for 1 minute to elute the DNA.5. RNase A Treatment (Optional)。

血液中DNA提取的原理:如果以外周血为DNA提取材料,在外周血中提取DNA就是从有核的白细胞中提取DNA。
因此,从外周血中提取DNA包括以下几个关键步骤:第一,破坏红细胞,通常利用红细胞与白细胞膜结构的差异,先使红细胞裂解,经离心后收集白细胞;第二,白细胞裂解:使膜蛋白和核蛋白变性,游离DNA,通常是采用离子型表面活性剂使蛋白质变性;第三,除去变性蛋白质:通常采用蛋白沉淀剂沉淀变性蛋白质,使DNA留在上清液中;第四,在高盐环境下使DNA从有机溶剂如无水乙醇中析出。
取材取外周血5ml,EDTA抗凝1.新鲜抗凝血,离心弃去上清液;2.取出4℃冰箱预冷的ELS裂解液,按1:6-10的比例向细胞沉淀中加入ELS裂解液(1ml 细胞压积加入6-10ml裂解液),轻轻吹打混匀;红细胞裂解液ELS配方:NH4Cl:4.15克加双蒸水500ml,取450ml;Tris:1.0297克加双蒸水50ml,再加上上面的450ml,共计500ml,高压灭菌,用时加10-20ml3.800-1000rpm离心5-8分钟,离心弃去上层红色清液;4.收集沉淀部分,加入Hank’s液或无血清培养液离心洗2-3次;5.如裂解不完全可重复步骤2和3;6.重悬细胞,用于后续实验;提取DNA,最好是于步骤4开始使用DEPC水配制的溶液进行.酚氯仿方法提取DNA在DNA提取过程中应尽量避免使DNA断裂和降解的各种因素,以保证DNA的完整性,为后续的实验打下基础。
是采用在EDTA以及SDS等试剂存在下用蛋白酶K消化细胞,随后用酚抽提而实现的。
这一方法获得的DNA不仅经酶切后可用于Southern分析,还可用于PCR的模板、文库构建等实验。
原则:(1)防止和抑制DNase对DNA的降解;(2)尽量减少对溶液中DNA的机械剪切破坏。
一、试剂准备1.TE: 10mM Tris-HCl (pH 7.8); 1mM EDTA (pH 8.0)。
2.TBS: 25mM Tris-HCl (pH 7.4); 200mM NaCl; 5mM KCl。
磁珠法提取游离dna以磁珠法提取游离DNA引言:DNA提取是生物学研究中的常用操作,它是分子生物学和遗传学研究的基础。
磁珠法提取游离DNA是一种高效、快速、准确的DNA 提取方法,广泛应用于基因组学、遗传学和临床医学等多个领域。
本文将介绍磁珠法提取游离DNA的原理、步骤和应用。
一、原理:磁珠法提取游离DNA的原理基于磁性珠子对DNA的亲和力。
磁珠是一种具有磁性的微小颗粒,表面经过修饰能够与DNA特异性结合。
在提取过程中,磁珠与DNA结合形成复合物,通过磁场的作用将复合物集中于一定位置,然后通过洗涤和离心等步骤去除杂质,最后用适当的缓冲液将DNA从磁珠上洗脱下来,得到纯净的游离DNA。
二、步骤:1. 样品处理:样品可以是血液、组织、细胞等含有DNA的生物材料。
首先,将样品进行预处理,如血液可以通过裂解红细胞膜释放DNA,组织可以经过细胞破碎等方法获得单个细胞。
2. 结合磁珠:将样品加入含有DNA结合磁珠的管子中,充分混合。
DNA在磁珠表面结合形成复合物。
3. 磁场分离:将含有复合物的管子放入磁场中,磁珠受磁力作用向管壁靠拢,使复合物沉积在管壁上。
废液中的杂质被去除,保留含有DNA的复合物。
4. 洗涤:加入适量的洗涤缓冲液,将其充分混合,然后将管子放入磁场中,使磁珠与洗涤液分离。
这一步的目的是去除残留的杂质,确保提取的DNA纯度。
5. 洗脱:加入适量的洗脱缓冲液,将其充分混合,然后将管子放入磁场中,使磁珠与洗脱液分离。
DNA从磁珠上洗脱下来,得到纯净的游离DNA。
三、应用:磁珠法提取游离DNA具有高效、快速、准确的特点,广泛应用于各个领域。
以下是一些常见的应用:1. 基因组学研究:磁珠法提取游离DNA是进行基因组DNA测序、SNP分型等研究的重要步骤。
2. 遗传学研究:磁珠法可用于提取个体DNA,用于遗传标记分析、基因突变检测等研究。
3. 临床医学:磁珠法提取游离DNA在临床诊断中有重要应用,如肿瘤基因检测、遗传病筛查等。
法医实验室几种常用DNA提取方法及比较DNA提取是法医学中常用的一项技术,用于获得从犯罪现场或是嫌疑人身上采集的样本中的DNA,从而用于分析和鉴定。
在法医实验室中,有几种常用的DNA提取方法,下面将对其进行介绍和比较。
1. 酚/氯仿提取法(Phenol/Chloroform Extraction Method):酚/氯仿提取法是DNA提取的经典方法之一、它通过将DNA样品与酚和氯仿等有机溶剂混合后,形成两相体系,利用酚的高密度和氯仿的高挥发性质,将DNA转移到有机相中。
通过离心分离DNA和有机溶剂,最终得到纯净的DNA。
该方法能够获得高纯度的DNA,适用于各种样本(血液、组织、唾液等),但操作复杂且耗时较长。
2. 快速提取法(Rapid Extraction Method):快速提取法是一种高效的DNA提取方法,适用于大规模样品的处理。
它利用一种化学试剂,如蛋白酶K,能够迅速裂解细胞并释放DNA。
然后通过简化的步骤,如磁珠吸附或粘附柱,将DNA从溶液中分离。
该方法操作简单、快速,适用于高通量的样本分析,如犯罪现场的大规模检材筛查。
3. 盐析法(Salting out Method):盐析法是一种常用的DNA提取方法,利用高盐浓度可以沉淀DNA。
首先,细胞样本经过溶解后,加入高盐浓度缓冲液,使DNA与其他细胞成分分离。
然后通过离心沉淀DNA,去除上清液。
最后,通过洗涤去除溶剂和杂质,获得纯净的DNA样品。
盐析法操作简单,适用于各类样本,但提取的DNA质量可能稍差。
4. 硅胶柱法(Silica Column Method):硅胶柱法是一种基于硅胶膜或胶珠的DNA提取方法。
这种方法通过将DNA样品与硅胶柱一起离心,使DNA吸附在硅胶上。
随后,通过洗涤去除杂质,最后用低盐缓冲液洗脱DNA,得到纯净的DNA。
硅胶柱法提取的DNA纯度和质量较高,适用于各种样品。
不过,硅胶柱价格较高,操作相对繁琐。
总体而言,这些DNA提取方法各有优劣,选择合适的方法需要考虑实验室的需求和条件。
TKM—血液DNA快速提取法
赵瑞君;赵仰星
【期刊名称】《山西医学院学报》
【年(卷),期】1996(027)001
【摘要】在临床PCR检验中,血卟啉对酶促反应的影响不容忽视,因此在纯化
DNA的过程中,常需将红细胞清除,以消除血卟啉对实验结果的干扰。
用TKM法提取血液DNA,方便、经济、快速且效果良好。
该DNA可用于PCR的分析,结果及为理想,完全可以替代其他血液DNA的提取方法。
【总页数】3页(P4-6)
【作者】赵瑞君;赵仰星
【作者单位】山西医学院寄生虫学教研室;山西省肿瘤医院血研室
【正文语种】中文
【中图分类】R446.9
【相关文献】
M法提取血液DNA调查山西地区人群的HUMTPOX基因座多态性 [J], 梁
景青;赵芳芳
2.石蜡包埋组织DNA快速提取法应用于分子病理检测价值评价 [J], 杨秀媚
3.适用于微卫星标记的湿地松、加勒比松DNA快速提取法 [J], 李义良;赵奋成;张应中;吴惠姗
4.一种适于转基因水稻PCR检测的微量DNA快速提取法 [J], 许明;程祖锌;黄志
伟;杨志坚;郑金贵
5.实验动物皮肤病原真菌DNA快速少量提取法 [J], 刘福英
因版权原因,仅展示原文概要,查看原文内容请购买。
Rapid Extraction of High Quality DNA from Whole Blood Stored at 4oC for Long Period
Materials and Methods
Standard chemicals
This method uses standard chemicals that can be obtained from any major supplier; we used
chemicals supplied by Sigma Co. as follow:
EDTA M), pH : Add gr of anhydrous EDTA to 800 ml of distilled water. Adjust pH to with
NaOH pellets. Make up to 1 liter with distilled water. Autoclave at 15 for 15 min.
1 M Tris-HC1, pH : Dissolve gr of Tris base in 800 ml of distilled water. Adjust pH with
concentrated HCl. Allow mixture to cool to room temperature before finally correcting pH. Make up to
1 liter with distilled water. Autoclave at 15 for 15 min.
Preparation of Red blood cell lysis buffer: M Tris-HCl pH , 320 mM sucrose, 5 mM MgC12, 1%
Triton X 100. Add 10 ml of 1 M Tris, gr of sucrose, gr MgC12, adjust pH to and finally add 10 ml of
Triton X-100 to 800 ml of distilled water, and make up to 1 liter with distilled water. Autoclave at 15
for 10 min. Sugars at high temperature can cause caramelization (browning), which degrades the sugars
[5].
Preparation of Nucleic lysis buffer: M Tris-HC1, mM sodium citrate, 1 mM EDTA, 1 % sodium
dodecyl sulphate (SDS). Take 10 ml of 1 M Tris-HC1 (pH , gr of anhydrous EDTA (pH , 10 gr SDS, gr of
sodium citrate, and adjust pH to . Make up to 1 liter with distilled water. Autoclave 15 min at 15
TE Buffer, pH : Take 5 ml of 1 M Tris-HCl, pH , 2 mL of M EDTA, pH 8, and make up to 1 liter with
distilled water. Adjust pH to and autoclave 15 min at 15.
Chloroform prechilled to 4°C.
Ethanol (100%) prechilled to -20°C.
Procedure of DNA Extraction
Before starting DNA extraction, liquid blood venogects should be shake gently by rotating blood
mixer (vortex)
1. Pour 500 μl of blood into a ml eppendorf tube and add 1000 μl of red cell lysis buffer.
2. Shake microfuge tube gently (up to homogenizing), then spin for 2 minutes at 7000 rpm.
3. Discard supernatant and repeat steps 1-3 two or three more times to remove hemoglobin. It is
important to breakdown the pellet by vortexing and rinses it well in red blood cell lysis buffer in order
to clean the white blood cells from residual of hemoglobin.
4. Placing the tube on tissue paper for few seconds downward. Be careful from cross-contamination
between different samples.
5. Add 400 μl of nucleic lysis buffer to eppendorf tube. Note: if the pellet formed, you must pipette
the pellet up to dissolve it.
6. Add 100 μl of saturated NaCl (5M) and 600 μl of chloroform to eppendorf tube and mix on a
rotating blood mixer at room temperature then spin it for 2 minutes at 7000 rpm.
7. Transfer 400 μl of supernatant to a new ml tube.
8. Add 800 μl of cold (-20°C) absolute Ethanol and shake it gently then vortex it. DNA should appear
as a mucus-like strand in the solution phase.
9. Spin the microfuge tube for one minute at 12000 rpm to precipitate, then discard supernatant
carefully and let tube be completely dried in room temperature (Place Eppendorf tube downward on
the tissue paper).
10. Add 50μl of TE to it then vortex; keep eppendorf tube of DNA in 4°C or -20°C for later uses. We
routinely use about one μl per PCR reaction without adverse affects. DNA can be quantified and diluted
to a working concentration at this point or simply use 1 μl per PCR reaction. We expect that the yield of
this procedure be 100 to 300 ng/μl, DNA. Using the above method, high quality DNA samples from a
sheep population were extracted for gene mapping studies.