微卫星详细实验流程
- 格式:pdf
- 大小:753.47 KB
- 文档页数:9

虎纹蛙基因组SSR标记的开发研究背景:简单序列重复(simple sequence repeats,SSRs)又称微卫星(microsatellites)标记具有共显性、高多态性、多等位性、稳定性好以及操作简单等诸多优点,作为理想的分子标记已广泛应用到人类、许多动物、植物的遗传学研究。
SSR 标记的开发有多种方法,包括传统的构建基因组文库、利用各种富集方法、基于ISSR-PCR 的方法、利用FIASCO 方法分离、借用近缘物种的标记以及较为简单的利用DNA 序列信息开发SSR 标记。
大量基因组和生物信息学资源不断产生,为快速开发生物SSR标记提供了机会。
虎纹蛙是中大型蛙科(Ranidae)动物,列入国家二级保护动物名录,国内主要见于长江以南地区的低海拔稻田、水塘和沟渠等地。
受人为捕捉、环境污染和生境破坏等影响,虎纹蛙野外数量已锐减,开展该种的基础研究显得尤为迫切。
此外泰国虎纹蛙大量引入和人工饲养,更是造成本地虎纹蛙种群数量的衰退。
本实验从虎纹蛙全基因组序列中发掘多态性SSRs,旨在为本地虎纹蛙物种的鉴定、遗传变异、系统进化、高密度分子作图和功能基因鉴定与克隆等研究提供丰富和更加有效SSR 标记。
研究方法:1.全基因组测序选取1个雄性虎纹蛙个体,取肌肉组织,进行DNA提取,并由北京诺和生物信息技术公司完成全基因组测序,DNA测序量(12G)约为总DNA量的10%。
2.DNA序列数据初步筛选采用SSR Hunter 1.3软件进行DNA序列数据中的SSR初筛。
重复元件的核苷酸数选为“4个”,重复次数最少为“5”。
从序列库中选取4000-10000行(每2行为1条DNA序列,包括DNA序列编号和碱基序列),粘贴于空白框内。
点击SSR 位点搜索,软件进行运算(基本数据量越大,越容易卡,导致软件显示“未响应”,(不用在意这个,其实软件一直在进行运算),一般10000行运行1-2小时不等),运算结束后,会弹出对话框,进行保存运算后的数据。




矿产资源开发利用方案编写内容要求及审查大纲
矿产资源开发利用方案编写内容要求及《矿产资源开发利用方案》审查大纲一、概述
㈠矿区位置、隶属关系和企业性质。
如为改扩建矿山, 应说明矿山现状、
特点及存在的主要问题。
㈡编制依据
(1简述项目前期工作进展情况及与有关方面对项目的意向性协议情况。
(2 列出开发利用方案编制所依据的主要基础性资料的名称。
如经储量管理部门认定的矿区地质勘探报告、选矿试验报告、加工利用试验报告、工程地质初评资料、矿区水文资料和供水资料等。
对改、扩建矿山应有生产实际资料, 如矿山总平面现状图、矿床开拓系统图、采场现状图和主要采选设备清单等。
二、矿产品需求现状和预测
㈠该矿产在国内需求情况和市场供应情况
1、矿产品现状及加工利用趋向。
2、国内近、远期的需求量及主要销向预测。
㈡产品价格分析
1、国内矿产品价格现状。
2、矿产品价格稳定性及变化趋势。
三、矿产资源概况
㈠矿区总体概况
1、矿区总体规划情况。
2、矿区矿产资源概况。
3、该设计与矿区总体开发的关系。
㈡该设计项目的资源概况
1、矿床地质及构造特征。
2、矿床开采技术条件及水文地质条件。
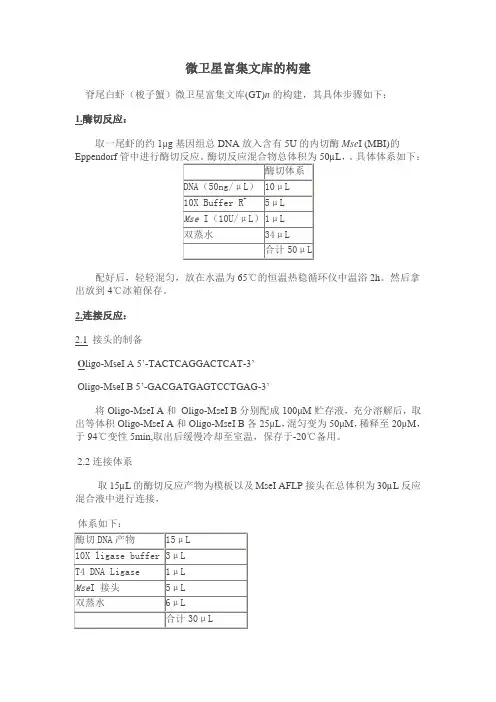
微卫星富集文库的构建脊尾白虾(梭子蟹)微卫星富集文库(GT)n的构建,其具体步骤如下:1.酶切反应:取一尾虾的约1μg基因组总DNA放入含有5U的内切酶Mse I (MBI)的Eppendorf50µL,。
具体体系如下:配好后,轻轻混匀,放在水温为65℃的恒温热稳循环仪中温浴2h。
然后拿出放到4℃冰箱保存。
2.连接反应:2.1 接头的制备O ligo-MseI A 5‟-TACTCAGGACTCAT-3‟Oligo-MseI B 5‟-GACGATGAGTCCTGAG-3‟将Oligo-MseI A和Oligo-MseI B分别配成100μM贮存液,充分溶解后,取出等体积Oligo-MseI A和Oligo-MseI B各25μL,混匀变为50μM,稀释至20μM,于94℃变性5min,取出后缓慢冷却至室温,保存于-20℃备用。
2.2连接体系取15µL的酶切反应产物为模板以及MseI AFLP接头在总体积为30µL反应混合液中进行连接,体系如下:混匀后,置22℃恒温水浴锅温浴1h,然后立即转到65℃水浴10min灭活T4 DNA Ligase。
连接产物保存在-20℃备用。
3. PCR预扩增:以上步酶切连接好的产物为模板,以Mse I-N:5‟-GATGAGTCCTGAGTAAN-3‟为引物进行PCR扩增,体系如下:PCR反应在MJ Research PTC-100 PCR仪上进行。
反应条件为:首先94℃预变性5min,然后在94℃变性30 s,53℃退火1 min,72℃延伸1 min;进行17个循环,最后72℃终延伸10min。
反应产物使用1%琼脂糖电泳检测,判断产物分布范围及产物量。
4. PCR预扩增产物的纯化使用普通DNA产物纯化试剂盒(TIANGEN),所有离心步骤均为使用台式离心机在室温下离心。
1 柱平衡:向吸附柱CB2中加入500μL的平衡液BL,13000rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
STR/SSR/微卫星分析背景介绍微卫星标记(Microsatellite)也称为短串联重复序列(STR)或简单重复序列(SSR), 是广泛分布在真核生物基因组中的简单重复序列。
微卫星位点通常通过PCR扩增和电泳检测, 并根据片段大小分离等位基因进行分析;扩增后的等位微卫星可以用多种方法检测, 传统方法采用聚丙烯酰胺凝胶电泳加放射显影或银染的方法, 费时费力效率低。
阅微基因基于5年的经验, 利用ABI遗传分析仪对荧光标记的DNA片段进行检测, 结合分子量内标进行DNA片段长度计算, 使STR分型变得高效快捷, 结果也更加精确。
服务项目1. SSR引物开发通过富集微卫星序列, 开发出20-30条具有一定重复特征的微卫星序列, 并且对序列进行多态性和特异性验证。
2.引物筛选选取部分代表性样本, 利用普通引物进行PCR扩增, 然后利用高分辨率芯片电泳或PAGE验证引物的特异性、扩增效率和基因座多态性等信息(图1)。
图1.用岛津MultiNA芯片电泳仪进行引物筛选的结果3.样本批量扩增用带有荧光标记(FAM/HEX/TMR/ROX等)的引物, 对批量样本进行PCR扩增。
我公司拥有高通量PCR仪平台, 可以满足大量样本扩增需求。
4.测序仪检测带有荧光标记的PCR产物进行稀释后与分子量内标混合, 用3730xl等测序仪进行毛细管电泳, 并得到原始数据(fsa文件, 见图2)。
图2.应用3730xl测序仪检测得到的原始图谱5. 原始数据分析编制panel和bin等文件, 使用正版GeneMapper v4.0软件分析测序仪得到的fsa文件, 并生成PDF图谱和Excel文档(包括size、基因分型等信息), 分析后的电泳图谱见图3。
图3.GeneMapper软件分析后的电泳图谱6. 群体遗传学分析利用生物信息学软件(POPGENE、MEGA.PHYLIP、ARLEQUIN等)对上述数据进行进一步分析, 绘制遗传连锁图谱、分析连锁不平衡和分析多态性等。

微卫星标记技术原理
微卫星标记技术是一种基于PCR (Polymerase Chain Reaction) 扩增的分子标记技术。
微卫星是基因组中较短、高度可变重复序列(Simple Sequence Repeats, SSRs),在不同个体或同一基因组中的
不同位置均有差异。
微卫星标记技术通过引物对位于微卫星序列的两
端的flanking序列进行PCR扩增,再以电泳方法进行分离和检测。
具体步骤包括:1. DNA提取,取得待检测组织的DNA;2. 引物
设计,设计与微卫星重复序列两侧的flanking序列可以特异性结合的
引物;3. PCR扩增,将待检测的DNA模板按设定条件进行PCR扩增;4. 电泳分离,将扩增产物以电泳方式分离出来;5. 图谱显示和分析,通
过显示分离出来的电泳带,分析出样品间和亲缘关系的差异以及基因
型信息。
微卫星标记技术具有高度多态性、重复性好、易于检测、特异性
强等优点。
它已广泛应用于动植物的遗传多样性、种间亲缘关系研究、基因定位及遗传距离测定等方面。

1.进行酶切用Vs pⅠ对四种鱼(GC、GS、NL和ZZ)基因组DNA(需基因组DNA浓度较高)进行酶切。
反应体系为:ddH2O 15µLVs pⅠbuffer 2µLVs pⅠ酶1µLDNA 2µL总体积20µL酶切在37℃反应3~4h即可,但是为了提高酶切效率可切4~6h。
PCR反应程序为:94℃ 5min,(94℃ 1min,53℃ 1min,72℃ 1min)×30,72℃ 10min,4℃ hold。
Vs pⅠ酶即(AseⅠ酶):酶切识别位点为:5’A T↓T A A T 3’5’T A A T↑T A 3’AseⅠ酶的接头为:A1:5’CTC GTA GAC TGC GTA CC 3’A2:5’TAG GTA CGC AGT C 3’AseⅠ酶用的预扩增产物为:A3:5’CTC GTA GAC TGC GTA CCT AAT 3’2.接头制备用10µL A1(200µM)和10µL A2(200µM)混合,94℃ 3min后室温放置30min。
3.酶切片段与接头连接接头为A1和A2制备的接头,命名为AE。
连接反应体系如下:ddH2O 5µLT4 ligation酶0.5µLT4 buffer4µL酶切产物20µLA TP(10mM)0.3µLAE 1µL总体积30.8µL上述混合体系在16℃连接过夜。
10h左右。
4.连接产物PCR扩增对连接产物进行PCR扩增,每个样本做4管。
扩增引物为A3。
反应体系如下:ddH2O 14µL10×buffer 2.5µLMg2+2µLdNTPs(10mM)0.5µLA3 1µLDNA(连接产物)5µLTaqE(2.5U/µL)0.2µL总体积25.2µLPCR反应程序为:(94℃ 30sec,56℃ 1min,72℃ 1min)×20,72℃ 10min,4℃ hold。

微卫星用来亲子鉴定的原理微卫星是一种短串联重复序列(short tandem repeat,STR)位点,由于其在基因组中具有多态性,因此被广泛应用于亲子鉴定领域。
亲子鉴定是通过比较目标个体与亲缘关系标本(通常是可能的亲生父母)的微卫星位点进行相似度分析,从而确定两者之间的关系。
微卫星的原理主要包括微卫星的检测和分析两个方面。
微卫星的检测是亲子鉴定的第一步,其方法主要有聚合酶链反应(PCR)和毛细管电泳。
首先,需要设计引物对微卫星位点进行PCR扩增。
引物是一对短的DNA片段,与目标微卫星序列的上下游区域互补。
在PCR反应中,引物与DNA模板结合,DNA聚合酶在引物的指导下将DNA序列复制扩增。
通过多轮PCR循环,可使微卫星序列的数量显著增加。
其次,PCR产物需要经过毛细管电泳分析。
电泳是一种将分子在电场中迁移的方法。
在毛细管电泳中,PCR产物被注入至玻璃毛细管中,并在电场的作用下在毛细管内迁移。
由于微卫星序列的长度不同,不同长度的PCR产物在电场下具有不同的迁移速度,从而形成一系列不同长度的峰。
通过检测这些峰的数量和大小,可以确定目标个体所携带的微卫星位点的等位基因型。
微卫星的分析是亲子鉴定的关键步骤,通过比较目标个体与亲缘关系标本的微卫星位点等位基因型来确定两者之间的关系。
在分析中,主要包括等位基因型的比较和计算相似度。
等位基因型的比较是通过将目标个体的等位基因型与亲缘关系参照样本的等位基因型进行比较来进行的。
亲缘关系参照样本通常是可能的亲生父母,他们的等位基因型通常已经确定。
通过比较,可以确定目标个体所携带的等位基因型是从哪一位亲缘关系参照样本中遗传的。
计算相似度是通过比较目标个体与亲缘关系参照样本在微卫星位点等位基因型上的一致性来进行的。
相似度通常通过比较两者等位基因型的共同位点来估计,共同位点的数量越多,相似度越高。
通过比较多个微卫星位点的相似度,可以得出最终的亲子鉴定结果。
总结来说,微卫星亲子鉴定的原理是通过PCR扩增和毛细管电泳分析目标个体与亲缘关系参照样本的微卫星位点等位基因型,并通过等位基因型的比较和相似度的计算来确定两者之间的关系。

实验步骤总结1 DNA提取按操作步骤来进行,需要注意的是:1,离心的时候注意盖住上面的那个盖子,离心时温度不用管无关紧要的;2,取样品时,量不要太多,尽量剪碎。
2 检测DNA是否提出电泳1,利用琼脂糖电泳,记住比例,一般检测DNA时比例是1%;意思就是1g的琼脂糖对应100ml的TAE,一般用40的电泳槽,也就是0.4g的琼脂糖。
EB是1:20的比例,40ul一般加2ul就可以了。
所以检测DNA时比例是EB2ul:琼脂糖0.4g:TAE缓冲液40。
2,如果TAE用完了,其配置的比例是用50x的,加20ml的50xTAE,再用蒸馏水配置到1000ml即可,相当于加980ml的蒸馏水。
3,注意加EB时用的枪头是T400的枪头,就是比较长的那个。
而点样或其他时候用的是T300的枪头。
4,点样时比例为1:5,及加1ul的溴酚蓝+5ul的样品;跑的电泳电压U=100v,I=100mA(也不一定,根据具体情况而定)。
3 测OD值,及检测DNA的浓度1,首先要校准,注意调试的方法,用blank那个;同时注意放的方向。
2,比例一般是1:69,意思就是加1体积的DNA,另外加69体积的蒸馏水即可;测的时候注意清洗每次。
3,每次的范围那个一般在1.8~2之间,如果<1.8一般是RNA,大于2的一般是蛋白质。
4,稀释到最后的单位为ng/ul。
4 DNA稀释已备PCR用1,首先学会计算,举例说明:测得的某一DNA浓度为5.1ug/ml,在DNA提取的最后一步是加了30体积TAE,而在检测DNA浓度时浪费了5体积,剩下25体积,可能有误差,则按20体积来算。
而测OD值时,稀释了70倍(即69体积水和1体积的DNA);我们以固定的50ng/ul为标准,则计算方法就是(5.1x70)/50=7倍,注意:这里可以近似计算就是约等于那种。
20体积那就是20x7=140;已经有20体积,则再加120体积就可以啦!这里有一点注意事项:假如用的是天根的DNA试剂提取盒,最后一步加的是30体积的TAE,而我用的那个来风的试剂盒最后加的是100体积的,所以不同的计量计算方法是不一样的,要根据具体用的哪一种试剂盒来定;现在规定来风试剂盒100体积的按照稀释到100还是按照天根那个来算。
微卫星DNA简单重复序(SSR)也称微卫星DNA,也可称为SSRP(Simple Sequence Repeat Polymorphisms),STMS (Sequence-tagged microsatellites)。
其串联重复的核心序列为1一6 bp,其中最常见是双核昔酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10一60个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
SSR具有以下一些优点:(l)一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA量少。
显然,在采用SSR技术分析微卫星DNA多态性时必须知道重复序列两端的DNA序列的信息。
如不能直接从DNA数据库查寻则首先必须对其进行测序。
1 微卫星DNA的构成及特点微卫星又称简单序列重复(Simple Se-quence Repeats,SSR)。
一般以1-6个碱基为核心序列,首尾相连组成的串联重复序列。
这种序列存在于几乎所有真核生物的基因组中,含量丰富,且呈随机均匀分布。
它们不仅大量分布于基因的间隔区和内含子中,而且还分布于基因的外显子和调控区(如启动子、增强子),真核生物平均每50-150 kb就存在一个微卫星位点,如在人类基因组中每6 kb就有1个微卫星,禽类基因组中约89 kb出现1个微卫星。
微卫星DNA数目巨大,人类基因组中约有5×104-1×105个(CA)n重复序列,重复次数一般为15-60次,重复单位相同,其长度一般小于200 bp,但也有的更长。
每个特定位点的微卫星DNA均由两部分构成:中间的核心区和外围的侧翼区。
1.进行酶切用Vs pⅠ对四种鱼(GC、GS、NL和ZZ)基因组DNA(需基因组DN A浓度较高)进行酶切。
反应体系为:d dH2O15µLVs pⅠbuffer 2µLVs pⅠ酶1µLDNA2µL总体积20µL酶切在37℃反应3~4h即可,但是为了提高酶切效率可切4~6h。
PCR反应程序为:94℃ 5min,(94℃ 1min,53℃ 1min,72℃ 1min)×30,72℃ 10min,4℃ hold。
Vs pⅠ酶即(AseⅠ酶):酶切识别位点为:5’A T↓T A A T 3’5’T A A T↑T A 3’AseⅠ酶的接头为:A1:5’CTC GTA GAC TGC GTA CC 3’A2:5’TAG GTA CGC AGT C 3’AseⅠ酶用的预扩增产物为:A3:5’CTC GTA GAC TGC GTA CCT AAT 3’2.接头制备用10µLA1(200µM)和10µLA2(200µM)混合,94℃ 3min后室温放置30min。
3.酶切片段与接头连接接头为A1和A2制备的接头,命名为AE。
连接反应体系如下:ddH2O5µLT4 ligati on酶0.5µLT4 buffer4µL酶切产物20µLA TP(10mM)0.3µLAE 1µL总体积30.8µL上述混合体系在16℃连接过夜。
10h左右。
4.连接产物PC R扩增对连接产物进行PCR扩增,每个样本做4管。
扩增引物为A3。
反应体系如下:ddH2O14µL10×buffer 2.5µLMg2+2µLdNTPs(10mM)0.5µLA3 1µLDNA(连接产物)5µLTaqE(2.5U/µL)0.2µL总体积25.2µLPCR反应程序为:(94℃ 30sec,56℃ 1min,72℃ 1min)×20,72℃ 10min,4℃ hold。
GeneMapper 软件微卫星分析中文简易操作手册微卫星分析流程:软件术语介绍:一.设定微卫星分析方法:1.登录GeneMapper软件:1.1选择开始 -- All Programs -- Applied Biosystems -- GeneMapper--GeneMapper ;或双击桌面快捷方式打开软件;1.2登录GeneMapper软件:用户名默认为gm,在Passwprd中输入密码,点击OK进入GeneMapper软件2创建分析所需kit、panel和marker等参数:2.1 点击软件上方工具栏Tools,选择Panel Manager;2.2 在Panel Manager对话框中,选中1 Panel Manager,点击2 新建 Kit;于New Kit对话框中依次输入Kit名称,Kit类型,并点击OK保存设置;2.3 在Panel Manager对话框中,选中1 STR KIT,点击2 新建 Panel;3中输入Panel名称;2.4 选中1 STR panel,点击2新建Marker,在3位置依次输入Marker Name(Marker名称) ,Dye Color (荧光标记种类) ,Min Size,Max Size (片段最小值和最大值) ,Marker Repeat (重复次数) ;2.5重复2.4步骤,完成所有Marker设置,点击OK保存;3. 创建新文件,导入数据:3 .1 点击1 新建文件夹,在2 Project Type中文件夹类型选择Microsatellite ,点击OK;3.2 导入数据:点击1 Add Samples To Project 添加数据,在弹出的对话框中选择待分析数据,通过3 Add To List 添加到右边的面板中,点击4 Add完成数据添加;4. 设置分析参数:4.1 完成数据添加后,在1 位置,Analysis Method中选择New Analysis Method新建分析方法;在弹出的对话框中选择分析类型2 Microsatellite,点击OK确认;4.2 设置分析参数:在Analysis Method Editor中,点击1 General,填写2 Name(分析方法名称);之后点击3 Allele,在将4 Bin Set选择none;其它参数为软件默认,不需要修改;点击OK确认;4.3 选定好之前设定的分析方法,在后续的栏目中设定Panel 和Size Standard(分子量内标);4.4 完成分析参数设定后,选中Analysis Method,Panel和Size Standard栏目,点击1File,选择2Fill Down;此时,Analysis Method,Panel和SizeStandard栏目会使用之前选定的分析参数;也可以使用鼠标分别选择;4.5 保存分析文件:点击File,选择Save Project,输入文件夹名称:5. 执行初始分析:5.1 点击分析按钮1,分析数据:5.2 在1 Genotypes中查看初步分析结果,或通过2 Display Plots选项查看样品分型图:5.3 在样品分型图界面中,Plot Setting选择 Microsatellite Default;通过软件上方的快捷按钮编辑数据显示方式:6 创建Bin Set:6.1 打开Tools—Panel Manager,选中1 STR Kit,点击2 新建Bin Set,在弹出的New Bin Set 对话框3中输入Bin Set名称:6.2 添加参考数据:6.2.1 确保1 Bin Set已选择新建的Bin Set,选中2 STR Panel,此时会显示出之前新建的每个Marker,点击3 Add Reference Data 添加参考数据;6.2.3 在Add Microsatellite Reference Data对话框中,选择1 参考数据,通过2Add To List 添加至右面的面板中,点击3 Add,完成添加:6.3 生成Bin Set:完成参考数据添加后,点击1 Auto Bin,之后使用软件默认参数,点击2 OK,自动生成Bin Set:6.4 编辑Bin Set:分别选中每个Marker 1 查看其对应的Bin,选中2 Bin,右键进行Bin的编辑或者删除,也可通过软件3处的快捷图标进行Bin的编辑;完成后点击OK生成Bin Set;二.分析并检查结果:1.编辑分析方法:点击软件上方快捷图标1 Analysis Method Editor,将Allele中的2 Bin Set修改为之前新建的Bin Set,其他参数保持默认,点击OK;2.点击分析按钮再次分析数据,通过Genotypes查看分型结果,或选中数据通过Display Plots查看样品分型图;选中需要查看的数据,否则无法查看Samples Plot:查看样品分型图:三.打印并导出数据(可选):1.在View中选择不同的界面(Samples,Genotypes等),再通过File—Export/Print 导出或者打印相应的数据:2.在Analysis 中选择不同的界面(Display Plots,Report Manager等),再通过File—Export/Print 导出或者打印相应的数据:。
ngs检测微卫星不稳定的原理
NGS(Next-Generation Sequencing,下一代测序)技术可以用
于检测微卫星不稳定性(microsatellite instability,MSI)。
微卫星不稳定性是由于DNA序列中的微卫星(短重复序列)
发生插入或缺失而导致的。
通常情况下,微卫星会经过DNA
修复系统的修复,使其保持稳定。
然而,在某些情况下,
DNA修复系统发生错误或缺陷,导致微卫星不稳定性的产生。
NGS技术可以通过同时测序多个DNA片段来检测微卫星不稳
定性。
具体步骤如下:
1. 样品制备:将DNA从病人或样品中提取出来。
2. 片段富集:使用特定的引物来扩增含有微卫星序列的DNA
片段。
3. 文库构建:将富集的DNA片段转化为一个可测序的文库。
4. 高通量测序:使用NGS技术对文库进行测序。
5. 数据分析:将测序数据与参考基因组进行比对,并检测微卫星序列的插入、缺失或变异。
6. 结果解读:根据微卫星序列的变异情况以及临床信息,确定微卫星不稳定性的状态。
NGS技术具有高通量、高分辨率和高灵敏度的特点,能够同时检测多个微卫星序列,提高检测效率和准确性。
因此,NGS技术在微卫星不稳定性的检测中被广泛应用。